
Abstract
Purpose of Review
This review provides an overview of the different aspects involved in the crosstalk between fibrogenesis, angiogenesis, and inflammation, contributing to liver disease progression.
Recent Findings
Fibrosis, characterized by abnormal and excessive deposition of extracellular matrix, results in compromised tissue and organ structure. This can lead to reduced organ function and eventual failure. Although activated hepatic stellate cells are considered the central players in fibrosis, the participation of other cell types and co-existing pathogenic processes to the initiation and progression of fibrosis has become increasingly recognized.
Summary
Understanding the pathophysiology of fibrosis and the molecular bases of hepatic stellate cell activation is essential to define novel and more efficient targets of antifibrotic therapy to reduce incidence, morbidity, and mortality of the people suffering from chronic liver disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic liver diseases are pathological processes that are characterized by the progressive destruction and regeneration of liver parenchyma resulting in the appearance of fibrosis and, in the longer term, cirrhosis [1••, 2].
A common trigger of the process, whatever its etiology (alcohol, viral infection, non-alcoholic steatohepatitis, or other), is liver damage that produces an inflammatory reaction. Continuous and repeated insults to the liver can lead to a scarring process with dysregulation of the synthesis and degradation of extracellular matrix resulting in its excessive deposition: fibrosis [1••, 2].
Chronic liver disease can progress to cirrhosis through increasing degrees of fibrosis, with alteration of the normal hepatic parenchymal architecture and formation of regenerative nodules surrounded by fibrous septa. This context favors the appearance of increasingly large hypoxic areas which, in turn, will promote vascular remodeling of the liver through angiogenesis. Angiogenesis and the formation of abnormal angioarchitecture have been widely described to be closely linked to progressive fibrosis and are considered major determinants of hepatic dysfunction and irreversibility in cirrhosis [3, 4].
This complex process involves all the cellular types of the liver, among which a series of relationships are established driving all the events described. The liver microenvironment, represented by the different cell types and the matrix scaffolding that supports them, becomes an interaction complex with a myriad of molecular signals, including autocrine, paracrine, endocrine, and cell–cell contacts that conduct the responses leading from liver damage to cirrhosis [5].
Cellular Crosstalk in the Liver
The particular structure of the liver sinusoid and its perisinusoidal space, the space of Disse, favors and promotes the communication between the different cell types of liver [6]. Thus, a crosstalk is established between hepatocytes, liver sinusoidal endothelial cells, hepatic stellate cells, Kupffer cells, and cholangiocytes, in both physiological and pathological situations. Through this crosstalk, a scenario is generated among the different cell types in which each one becomes the regulator of the others by means of a variety of growth factors, cytokines, and other signals. For example, the secretion of vascular endothelial growth factor (VEGF) by the hepatocytes contributes to the maintenance of the normal phenotype of the liver sinusoidal endothelial cells or the production of reactive oxygen species and proinflammatory cytokines by hepatocytes, Kupffer cells, or sinusoidal endothelial cells in response to cell damage. This promotes the activation of hepatic stellate cells and their differentiation into a myofibroblastic phenotype [1••, 5, 7,8,9].
A large number of ligand-receptor relationships have been described between liver cells, a fact that reflects the complex crosstalk that occurs in this organ, whether in healthy or pathological conditions [10••]. A recent study by Xiong and collaborators developed a global map of secretome gene expression in the liver of mice with non-alcoholic steatohepatitis. This map revealed that, in addition to the known crosstalk through factors secreted by resident cells, ligands of extrahepatic origin may play very important roles in the regulation of the biology of different hepatic cell types, since numerous endocrine and neuroendocrine receptors were detected in non-parenchymal cells of the liver [8].
Recent studies also point to extracellular vesicles as critical mediators of cellular crosstalk in the diseased liver [11, 12]. As an example, Witek and collaborators found that hepatic stellate cells and cholangiocytes release exosomes containing Hedgehog ligand to liver sinusoidal endothelial cells leading to endothelial dysfunction in cirrhosis [13].
In summary, the different cell types of the liver, whether in physiological or pathological conditions, establish a complex and multidirectional cell crosstalk that allows each one to influence the others, a fact that explains the relationship between the different processes involved in the progression of chronic liver disease [14, 15].
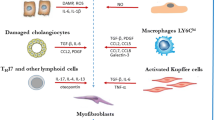
In this review, we discuss each of the components of the process from liver damage to fibrosis and cirrhosis (inflammation, fibrogenesis, hypoxia, angiogenesis), focusing on how each one is related to the others, paying special attention to the close relationship between angiogenesis and fibrosis (Fig. 1).

Link between angiogenesis, inflammation, and fibrosis in chronic liver disease. ECM, extracellular matrix; HSC, hepatic stellate cells; MMP9, matrix metalloproteinase-9; PAF, platelet-activating factor; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor
Inflammation
Inflammation in response to liver damage is a common trigger of many chronic liver diseases and an important driver of the progression from damage to fibrosis and cirrhosis. Different cell types in the liver, including Kupffer cells or hepatic stellate cells, respond to acute or chronic insults by releasing proinflammatory cytokines to kick-start the healing process after tissue damage and to recruit circulating inflammatory cells. Activated Kupffer cells (liver-resident macrophages) secrete, in addition to inflammatory cytokines, reactive oxygen species and factors such as platelet-activating factor, which are strong activators of angiogenesis [16, 17].
Targeting macrophages to treat liver disease has become one common strategy in many recent studies. The recruitment of monocyte-derived macrophages is a well-known mechanism of hepatic inflammation perpetuation and fibrogenesis promotion through activation of hepatic stellate cells, in response to secretion of proinflammatory cytokines by the recruited macrophages [10••, 18, 19]. Ehling and colleagues demonstrated that when inhibiting monocyte infiltration to the liver upon induction of hepatic damage, angiogenesis but not fibrogenesis was attenuated. This study showed that infiltrating CCL2-dependent inflammatory monocytes also provide proangiogenic signals, such as the production of VEGF-A or matrix metalloproteinase-9 (MMP9), which mediate fibrosis-associated angiogenesis. According to these results, inflammation-associated angiogenesis is thought to be involved in the earlier stages of fibrosis [18].
Toll-like receptor 4 (TLR4) signaling pathway activation by bacterial endotoxin (lipopolysaccharide) plays an important role in chronic and acute inflammatory disorders. It is expressed in several cell types of the liver and one of its functions is to mediate the production of proinflammatory cytokines. It has been well characterized in Kupffer cells and hepatic stellate cells [20••], but a study by Jagavelu and collaborators focused in the role of TLR4 in the liver endothelial cells. By inhibiting TLR4 signaling in mouse models of liver fibrosis, the authors observed a decrease in angiogenesis in parallel with fibrosis, providing a link between both processes mediated by TLR4 signaling in endothelial cells [21].
Fibrogenesis
As already mentioned above, liver fibrosis is described as the excessive deposition of fibrillar extracellular matrix as a consequence of persistent inflammation and sustained liver damage that leads to an exaggerated process of tissue repair with the main goal of preserving the continuity of the tissue. This accumulation of fibers delimits regenerative parenchyma nodules, isolated areas of liver tissue, altering the normal architecture and functions of the organ. It occurs in most types of chronic liver diseases and represents the initial steps in the process that leads to cirrhosis and, eventually, hepatocellular carcinoma [22].
Hepatic stellate cells are the liver-specific pericytes playing a key role in the initiation, progression, and regression of liver fibrosis. These cells represent between 5 and 8% of the total liver cells. They are located in the subendothelial space of Disse, defined as the space between the liver sinusoidal endothelial cells and the parenchymal cells. In a healthy adult liver, the main functions of quiescent liver pericytes are storage of retinols or vitamin A in cytoplasmic lipid droplet, regulation of sinusoidal blood flow (with their cytoplasmic extensions located along and around the sinusoids), regulation of the extracellular matrix turnover, and secretion of a variety of cytokines and growth factors. In situations of liver injury, hepatic stellate cells undergo a process of activation through which they release vitamin A and acquire myofibroblastic phenotype characterized by high proliferative, synthetic, and contractile capacity. Although there are other sources of myofibroblasts in the liver, such as portal fibroblasts or recruited bone marrow–derived mesenchymal cells, the activation of hepatic stellate cells is considered the main one [1••].
Although hepatic stellate cell activation constitutes one fundamental step of the development of hepatic fibrosis, other non-parenchymal cell types in the liver, such as liver sinusoidal endothelial cells or Kupffer cells and other inflammatory cells (resident or recruited upon liver damage), also play an important role in the regulation of fibrogenesis. Thus, the response of the liver to damage is orchestrated through the coordinated exchange of stimuli between all its different cell types.
A key point in the study of the relationship between fibrosis and angiogenesis is the proangiogenic activity of activated hepatic stellate cells and other myofibroblasts and their ability to respond to proangiogenic cytokines. The close anatomic relationship between hepatic stellate cells and sinusoidal endothelial cells, and the fact that stellate cells secrete proangiogenic factors upon their activation, points to a key role of this cell type in angiogenesis induction in association with fibrosis progression [23, 24].
As previously commented, hepatic stellate cell contribution to hepatic angiogenesis is not limited to stabilize and allow new blood vessel maturation, according to their role as microcapillary pericytes. Activated stellate cells have been shown to increase VEGF expression during activation [25,26,27,28]. In addition, hepatic stellate cells also increase the expression of the VEGF type I and type II (Flt-1 and Flk-1) receptors and the Tie-2 receptor for angiopoietin in conditions of hypoxia [3, 28, 29]. On the other hand, the upregulation of VEGF in response to hypoxia in hepatic stellate cells can stimulate migration, proliferation, and chemotaxis of the stellate cells themselves in an autocrine or paracrine way [3, 4, 29, 30]. This fact can explain the antifibrotic effects of the blockage of the VEGF signaling pathway. Furthermore, a study by Taura and collaborators highlighted the interrelationship between the different processes present in the progression of liver disease, mediated by hepatic stellate cells. The study showed that in response to hypoxia and through HIF-1α transcription factor, hepatic stellate cells overexpress VEGF and angiopoietin. Data obtained from human and murine fibrotic and cirrhotic livers found the presence of myofibroblastic phenotype cells at the edges of incomplete fibrous septa under development, which expressed VEGF and Ang-1. The blockade of Ang-1 signaling resulted in suppression of angiogenesis, paired to a reduction in fibrosis development, pointing at angiogenesis as a prerequisite for fibrosis in this model [24].
Hypoxia
The changes in the normal architecture of the hepatic parenchyma caused by the fibrogenic response to liver damage, with formation of regenerative nodules surrounded by fibrous septa, lead to a greater contribution of the hepatic artery to the sinusoidal circulation. This arterialization results in the capillarization of the sinusoids, and thus, the unique phenotype of the sinusoidal endothelial cells is lost. The progressive decrease of fenestrations and the formation of basement membrane in these cells, together with the activation of the hepatic stellate cells, lead to an increase in vascular resistance and the consequent deficit of oxygen transport to the hepatic parenchyma. This hypoxic environment, as previously mentioned, promotes angiogenesis and vascular remodeling of the liver. Thus, a feedback loop is established between fibrosis and pathological angiogenesis with hypoxia as the driving force [3, 4, 29,30,31,32].
Despite the fact that hypoxia-induced angiogenesis forms new vessels in an attempt to reoxygenate the hypoxic areas of the liver, these vessels are immature and poorly functional so they can hardly alleviate hypoxia. Persistent hypoxia accompanied by angiogenesis, inflammation, and fibrogenesis enters a vicious circle that leads to exacerbation of the disease [3].
Hypoxia and the hypoxia-inducible factors (HIFs) play an important role in chronic liver disease. Many studies have described the association of hypoxia with the parallel progression of angiogenesis and fibrogenesis, since progressively more hypoxic areas express higher levels of VEGF, to which not only endothelial cells respond but also other cell types, such as hepatic stellate cells or hepatocytes [3, 33,34,35]. Moreover, numerous studies have reported vascularization and/or the presence of endothelial cells within fibrotic structures [33,34,35,36]. A study conducted by Moon and collaborators demonstrated that HIF-1α-deficient mice developed less fibrosis. Using a model of secondary biliary cirrhosis, the authors pointed that the appearance of hypoxic areas in the liver as a result of liver damage promotes the activation of HIF. The perpetuation of this activation leads to continuous production of growth factors that ultimately stimulate the overproduction of extracellular matrix and fibrosis [31].
Angiogenesis
Angiogenesis is a ubiquitous process whose purpose is the formation of new blood vessels from the preexistent vasculature [37]. It is mainly stimulated by hypoxia and driven by growth factors. Angiogenesis occurs either in physiological conditions or in many different pathophysiological scenarios, including chronic liver disease [33, 38,39,40].
In chronic liver diseases, which are characterized by persistent activation of wound healing responses, fibrogenesis, hypoxia, and inflammation, angiogenesis plays a key role in the intrahepatic vascular remodeling. This remodeling affects the two differentiated vascular beds present in the liver: sinusoids, lined by a fenestrated endothelium of sinusoidal endothelial cells that facilitates efficient hepatocyte perfusion, and larger vessels, with continuous endothelial coating. In liver disease, hepatic sinusoids are modified by a capillarization process through which sinusoidal endothelial cells change their phenotype leading to endothelial dysfunction, with deposition of a basement membrane and loss of fenestrae [14]. The new vessels resulting of angiogenic processes originate mostly in the branches of the portal vein and have the purpose of establishing connections between the portal venous system and the hepatic veins [41].
There are two main sources of proangiogenic stimulation that explain the angiogenic processes in the liver in the context of chronic liver disease. First, the common wound healing response in these pathologies is characterized by the enormous production of growth factors and chemokines, such as PDGF, FGF, VEGF, or TGF-β1, that have strong proangiogenic capacity [42,43,44]. Second, hypoxia, the main stimulus for angiogenesis, is closely linked to the development of changes in the structure of liver parenchyma in chronic liver disease. Therefore, the growing areas of hypoxic tissue promote the formation of new blood vessels in the liver [3, 36, 41, 45].
In the process of neovascularization, VEGF plays a predominant role in the initial stages of formation of new blood vessels, activating the proliferation of endothelial cells and the subsequent formation of an endothelial tubule, while maturation of the newly formed vessels is mainly modulated by the proangiogenic growth factor platelet-derived growth factor (PDGF), which regulates the investiture of the endothelial tubule with pericytes, thereby stabilizing the vascular architecture of the nascent vessel. Based on these considerations, it was hypothesized that the simultaneous targeting of the VEGF and PDGF signaling pathways, that is, the simultaneous targeting of endothelial cells and pericytes, could provide a greater vascular destabilization and a better vascular regression than targeting either alone. Indeed, combined antiangiogenic treatment directed against endothelial cells and pericytes, using VEGF and PDGF inhibitors simultaneously, provides a synergistic benefit in reducing circulatory abnormalities in portal hypertension [34]. This is due to the fact that removal of pericyte coverage by anti-PDGF molecules leads to exposed endothelial tubes, making endothelial cells much more susceptible to anti-VEGF treatment. In this regard, concurrent targeting of VEGFR-2 and PDGFR-β signaling pathways using low doses of the multikinase inhibitor sorafenib significantly reduces portosystemic collateralization, hyperdynamic splanchnic circulation, intrahepatic fibrosis, and portal pressure in experiments in rats with cirrhosis [34]. These findings further suggest that in the absence of proliferating pericytes (i.e., after PDGF signaling inhibition), the endothelium is more vulnerable to antiangiogenic therapies targeting endothelial cells, such as VEGF signaling blockade.
Angiogenesis, Hand in Hand with Fibrogenesis
A great number of studies from several groups point at a connection between angiogenesis and fibrogenesis in the progression of chronic liver diseases. Both processes develop in parallel in different human chronic liver diseases and animal models [46,47,48,49,50]. The progression of fibrogenesis leading to cirrhosis is closely linked to the formation of new blood vessels (angiogenesis) and to the disturbance of the normal hepatic vascular structure. Aberrant angiogenesis is undoubtedly implicated in the progression of hepatic fibrosis and is considered a major determinant of hepatic dysfunction and irreversibility in cirrhosis [51].
Supporting this statement, many different antiangiogenic strategies have been proved effective in reducing fibrogenesis in experimental models [52]. Wang and collaborators described the anti-fibrogenic potential in vivo and in vitro of TNP-470, a semisynthetic analogue of fumagillin used as an angiogenesis inhibitor. Their results showed that TNP-470 administration translated into a decrease of hepatic stellate cell activation and proliferation, reducing the progression of hepatic fibrosis, probably due to its antiangiogenic effect [53].
Other studies have focused in VEGF and its receptors, since this is one of the most important proangiogenic signaling pathways. Using neutralizing monoclonal antibodies targeting VEGFR1 and VEGFR2, Yoshiji and collaborators demonstrated that by blocking the union of VEGF to its receptors (especially VEGFR2), fibrosis development was significantly reduced in the CCl4-induced mouse model of fibrosis [23]. Using a similar strategy, it was shown that bevacizumab, a monoclonal antibody capable of neutralizing all isoforms of VEGF-A, inhibits hepatic stellate cell activation and attenuates fibrosis in the CCl4-induced rat model of fibrosis [54].
Other authors have used multikinase inhibitors to influence other proangiogenic signaling pathways in addition to VEGF. It is the case of sunitinib or sorafenib [55,56,57,58,59]. Both bind to the binding sites of their target kinases inhibiting their catalytic activity. Sunitinib (SU11248) treatment on cirrhotic rats resulted in a decrease of inflammatory infiltrate to the liver, reduction of hepatic vascularization, and attenuation of the fibrogenic response. These effects were accompanied by downregulation of alpha-smooth muscle actin and collagen expression in vivo and reduction of the viability of LX2 cells (a human hepatic stellate cell line) in vitro [55, 56]. Similarly, the treatment of common bile duct–ligated rats with sorafenib, which potently blocks the tyrosine kinases of VEGF receptor-2 (VEGFR-2) and PDGF receptor-β (PDGFR-β), as well as the Raf serine/threonine kinases along the Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal–regulated kinase (ERK) pathway, resulted in a remarkable improvement in liver damage and intrahepatic fibrosis, inflammation, and angiogenesis [57]. A subsequent study used another fibrosis model in rats (dimethyl nitrosamine injection) to test the effects of sorafenib, as well as in vitro on LX-2 cells. This study confirmed the potential of sorafenib as a therapeutic agent in the treatment of liver fibrosis, since the treatment with this drug resulted in the suppression of collagen accumulation, reduction of hepatic stellate cell proliferation, and induction of their apoptosis [59].
Moreover, two recent studies investigated the effects of the overexpression of endogenous inhibitors of angiogenesis (pigment epithelium–derived factor (PEDF) and vasohibin-1) by adenovirus-mediated gene transfer to bile duct–ligated rats. Both studies observed a reduction of pathologic angiogenesis accompanied by an attenuation of liver fibrosis by increasing the levels of these endogenous inhibitors of angiogenesis. The mechanisms underlying the reduction of fibrosis were different in both cases: PEDF affected the expression of metalloproteinases whereas vasohibin-1 partially inhibited the activation hepatic stellate cells [60, 61].
Despite the beneficial effects found in many studies, the inhibition of angiogenesis as a therapeutic strategy in the context of chronic liver diseases should be faced with extreme caution. This is due to the fact that VEGF plays a dual role in liver fibrosis, since it contributes not only to fibrogenesis but also to fibrolysis or fibrosis resolution. Yang and collaborators demonstrated in a model of common bile duct–ligated mice that VEGF promoted fibrogenesis, but was also required for hepatic tissue repair and fibrosis resolution. This study demonstrated that VEGF signaling inhibition using VEGF-neutralizing antibodies attenuated development of fibrosis, but on the other hand, it also impaired fibrosis resolution and liver repair, since the lack of VEGF signaling resulted in impaired permeability of liver sinusoidal endothelial cells, which decreased infiltration of monocytes [62]. In addition, VEGF contributes to the maintenance of the normal phenotype of liver sinusoidal endothelial cells, which, in turn, prevents the activation of hepatic stellate cells and promotes their return to the quiescent state once activated [63]. Furthermore, macrophages infiltrating into fibrotic areas have also been proved to play a dual role in fibrosis promotion and resolution. Therefore, although these macrophages stimulate fibrogenesis in the early stages, it has been found that myeloid cell–derived VEGF drives a pro-resolution phenotype in liver sinusoidal endothelial cells that facilitates the revascularization of fibrous tissue during fibrosis resolution [64].
Posttranscriptional Reprogramming in Angiogenesis and Fibrogenesis
The observations described above emphasize the importance of deciphering mechanisms that specifically regulate pathological angiogenesis without affecting the normal physiological vascularization. In this regard, we have recently identified a new mechanism of regulation of pathologic VEGF expression and angiogenesis in chronic liver disease, through sequential and nonredundant functions of two members of the family of cytoplasmic polyadenylation element binding proteins, CPEB1 and CPEB4 (Fig. 2) [65]. CPEB proteins are RNA-binding proteins that bind to and regulate the expression of a specific group of mRNAs, which have, on their noncoding 3′-untranslated regions (3′UTR), some sequences named cytoplasmic polyadenylation elements (CPEs) [66••, 67,68,69]. Upon cirrhosis induction, there is a rapid upregulation and activation by autophosphorylation of the serine/threonine kinase Aurora kinase-A in splanchnic organs and the liver. Activated Aurora kinase-A, in turn, phosphorylates and activates CPEB1 [70, 71]. Activation of CPEB1 then promotes alternative nuclear processing within 3′UTRs of VEGF and CPEB4 mRNAs, resulting in deletion of translation repressor elements. The subsequent overexpression of CPEB4 promotes cytoplasmic polyadenylation of VEGF mRNA, increasing its translation and generating high levels of VEGF [65]. Importantly, this CPEB-mediated regulatory mechanism is essential for pathologic angiogenesis but dispensable for physiologic neovascularization. Thus, targeting CPEBs could lead to safer treatment outcomes by specifically reducing excessive pathologic VEGF production instead of indiscriminately perturbing both pathologic and physiologic VEGF synthesis, minimizing potential adverse side effects [65]. More recently, we have identified a new facet of the “proangiogenic” activity of CPEB4. Thus, we have found that CPEB4 is required for the proliferation of progenitor stem cells found in the vascular wall of blood vessels, which are able to differentiate and mediate the abnormal growth of new vessels during liver disease [72]. Together, these findings reinforce the potential of CPEB4 as a therapeutic target for chronic liver disease and potentially also other angiogenesis-dependent diseases such as cancer [73••].

The RNA-binding proteins CPEBs are essential regulators of pathological angiogenesis and fibrogenesis in chronic liver disease. CPEB, cytoplasmic polyadenylation element binding protein; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3; VEGF, vascular endothelial growth factor
Interestingly, we have recently unveiled a previously unrecognized posttranscriptional regulatory circuit in activated hepatic stellate cells, comprising the critical glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) and the RNA-binding protein CPEB4 [74]. This circuit maintains hepatic stellate cells in fibrotic livers with a higher glycolytic activity than normal stellate cells in healthy livers. This abnormal glycolytic reprogramming predisposes hepatic stellate cells to activation, myofibroblast transdifferentiation, cellular proliferation, and extracellular matrix production and accumulation, resulting in exponentation of liver fibrosis. These findings further support the relevance of CPEB4 as a potential druggable target for chronic liver disease (Fig. 2).
Conclusions
In summary, in the context of chronic liver disease, persistent hepatic damage and inflammation are associated with mutual aggravation of fibrosis and hypoxia. Additionally, tissue hypoxia represents the main driving force promoting the initiation of angiogenesis and fibrogenesis in the liver. In this scenario, the extent of angiogenesis is a key factor in the rate of progression of fibrosis to cirrhosis and in the reversibility of fibrosis. This fact motivates that the association between fibrogenesis and angiogenesis should be considered key in the evaluation of disease progression and in the search for therapeutic strategies.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
•• Tsuchida, T; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017, 14, 397–411 Excellent review highlighting the remarkable complexity and plasticity of hepatic stellate cell activation.
Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Dig Liver Dis. 2004;36:231–42.
Rosmorduc O, Wendum D, Corpechot C, Galy B, Sebbagh N, Raleigh J, et al. Hepatocellular hypoxia-induced vascular endothelial growth factor expression and angiogenesis in experimental biliary cirrhosis. Am J Pathol. 1999;155:1065–73.
Paternostro C, David E, Novo E, Parola M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J Gastroenterol. 2010;16:281–8.
Sato K, Kennedy L, Liangpunsakul S, Kusumanchi P, Yang Z, Meng F, et al. Intercellular communication between hepatic cells in liver diseases. Int J Mol Sci. 2019;20:2180.
Wisse E, Braet F, Luo D, De Zanger R, Jans D, Crabbé E, et al. Structure and function of sinusoidal lining cells in the liver. Toxicol Pathol. 1996;24:100–11.
Thomson J, Hargrove L, Kennedy L, Demieville J, Francis H. Cellular crosstalk during cholestatic liver injury. Liver Res. 2017;1:26–33.
Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; Zhou, L.; Chen, Z.; Leon-Mimila, P.; Chung, M.T.; Kurabayashi, K.; Opp, J.; Campos-Pérez, F.; Villamil-Ramírez, H.; Canizales-Quinteros, S.; Lyons, R.; Lumeng, C.N.; Zhou, B.; Qi, L.; Huertas-Vazquez, A.; Lusis, A.J.; Xu, X.Z.S.; Li, S.; Yu, Y.; Li, J.Z.; Lin, J.D. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell 2019, 75, 644–660.
Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–30.
•• Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 2017, 17, 306–321 Excellent review highlighting novel findings regarding the origin, classification, and function of hepatic macrophages in healthy and diseased liver.
Cai S, Cheng X, Pan X, Li J. Emerging role of exosomes in liver physiology and pathology. Hepatol Res. 2017;47:194–203.
Moran L, Cubero FJ. Extracellular vesicles in liver disease and beyond. World J Gastroenterol. 2018;24:4519–26.
Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, et al. Liver cell-derived microparticles activate Hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–30.
Greuter T, Shah VH. Hepatic sinusoids in liver injury, inflammation, and fibrosis: new pathophysiological insights. J Gastroenterol. 2016;51:511–9.
Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. 2018;15:555–67.
Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. 2017;8:2247.
Li P, He K, Li J, Liu Z, Gong J. The role of Kupffer cells in hepatic diseases. Mol Immunol. 2017;85:222–9.
Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 1960-1971;2014:63.
Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300–12.
•• Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007, 13, 1324–1332 Excellent work demonstrating that TLR4 drives myofibroblast activation and fibrogenesis in the liver and that TLR4-dependent modulation of TGF-beta signaling provides a link between proinflammatory and bava and profibrogenic signals.
Jagavelu K, Routray C, Shergill U, O’Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology. 2010;52:590–601.
Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56:769–75.
Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut. 2003;52:1347–54.
Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 1729-1738;2008:135.
Mashiba, S.; Mochida, S.; Ishikawa, K.; , Inao, M.; Matsui, A.; Ohno, A.; Ikeda, H.; Nagoshi, S.; Shibuya, M.; Fujiwara, K. Inhibition of hepatic stellate cell contraction during activation in vitro by vascular endothelial growth factor in association with upregulation of FLT tyrosine kinase receptor family, FLT-1. Biochem Biophys Res Commun 1999, 258, 674–678.
Ishikawa K, Mochida S, Mashiba S, Inao M, Matsui A, Ikeda H, et al. Expressions of vascular endothelial growth factor in nonparenchymal as well as parenchymal cells in rat liver after necrosis. Biochem Biophys Res Commun. 1999;254:587–93.
Ankoma-Sey V, Wang Y, Dai Z. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology. 2000;31:141–8.
Ankoma-Sey V, Matli M, Chang KB, Lalazar A, Donner DB, Wong L, et al. Coordinated induction of VEGF receptors in mesenchymal cell types during rat hepatic wound healing. Oncogene. 1998;17:115–21.
Novo, E.; Cannito, S.; Zamara, E.; Valfrè di Bonzo, L.; Caligiuri, A.; Cravanzola, C.; Compagnone, A.; Colombatto, S.; Marra, F.; Pinzani, M.; Parola, M. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol 2007, 170, 1942–1953.
Copple BL, Bai S, Burgoon LD, Moon JO. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int. 2011;31:230–44.
Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1 alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–92.
Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010;30:258–70.
Medina J, Arroyo AG, Sánchez-Madrid F, Moreno-Otero R. Angiogenesis in chronic inflammatory liver disease. Hepatology. 2004;39:1185–95.
Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–20.
DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis Hepatology 2015, 61, 1740-1746.
Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–21.
Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4.
Garcia-Monzon C, Sanchez-Madrid F, Garcia-Buey L, Garcia-Arroyo A, Garcia-Sanchez A, Moreno-Otero R. Vascular adhesion molecule expression in viral chronic hepatitis: evidence of neoangiogenesis in portal tracts. Gastroenterology. 1995;108:231–41.
Medina J, Sanz-Cameno P, Garcia-Buey L, Martin-Vilchez S, Lopez-Cabrera M, Moreno-Otero R. Evidence of angiogenesis in primary biliary cirrhosis: an immunohistochemical descriptive study. J Hepatol. 2005;42:124–31.
Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011;31:146–62.
Hoofring A, Boitnott J, Torbenson M. Three-dimensional reconstruction of hepatic bridging fibrosis in chronic hepatitis C viral infection. J Hepatol. 2003;5:738–41.
Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416.
Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–76.
Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Insights into the pathobiology of hepatitis C virus-associated cirrhosis: analysis of intrahepatic differential gene expression. Am J Pathol. 2002;160:641–54.
DeLeve LD. Hepatic microvasculature in liver injury. Semin Liver Dis. 2007;27:390–400.
Elpek GO. Angiogenesis and liver fibrosis. World J Hepatol. 2015;7:377–91.
Park S, Kim JW, Kim JH, Lim CW, Kim B. Differential roles of angiogenesis in the induction of fibrogenesis and the resolution of fibrosis in liver. Biol Pharm Bull. 2015;38:980–5.
Bocca C, Novo E, Miglietta A, Parola M. Angiogenesis and fibrogenesis in chronic liver diseases. Cell Mol Gastroenterol Hepatol. 2015;13:477–88.
Zhang Z, Zhang F, Lu Y, Zheng S. Update on implications and mechanisms of angiogenesis in liver fibrosis. Hepatol Res. 2015;45:162–78.
Valfre di Bonzo, L.; Novo, E.; Cannito, S.; Busletta, C.; Paternostro, C.; Povero, D.; Parola, M. Angiogenesis and liver fibrogenesis. Histol Histopathol 2009, 24, 1323–1341.
Parola M, Marra F, Pinzani M. Myofibroblast-like cells and liver fibrogenesis: emerging concepts in a rapidly moving scenario. Mol Asp Med. 2008;29:58–66.
Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 1887-1901;2013:123.
Wang YQ, Ikeda K, Ikebe T, Hirakawa K, Sowa M, Nakatani K, et al. Inhibition of hepatic stellate cell proliferation and activation by the semisynthetic analogue of fumagillin TNP-470 in rats. Hepatology. 2000;32:980–9.
Huang Y, Feng H, Kan T, Huang B, Zhang M, Li Y, et al. Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS One. 2013;8:e73492.
Tugues S, Fernandez-Varo G, Muñoz-Luque J, Ros J, Arroyo V, Rodés J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology. 1919-1926;2007:46.
Majumder S, Piguet AC, Dufour JF, Chatterjee S. Study of the cellular mechanism of sunitinib mediated inactivation of activated hepatic stellate cells and its implications in angiogenesis. Eur J Pharmacol. 2013;705:86–95.
Mejias M, Garcia-Pras E, Tiani C, Miquel R, Bosch J, Fernandez M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology. 2009;49:1245–56.
Hong F, Chou H, Fiel MI, Friedman SL. Antifibrotic activity of sorafenib in experimental hepatic fibrosis: refinement of inhibitory targets, dosing and window of efficacy in vivo. Dig Dis Sci. 2013;58:257–64.
Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–44.
Mejias M, Coch L, Berzigotti A, Garcia-Pras E, Gallego J, Bosch J, et al. Antiangiogenic and antifibrogenic activity of pigment epithelium-derived factor (PEDF) in bile duct-ligated portal hypertensive rats. Gut. 2015;64:657–66.
Coch L, Mejias M, Berzigotti A, Garcia-Pras E, Gallego J, Bosch J, et al. Disruption of negative feedback loop between vasohibin-1 and vascular endothelial growth factor decreases portal pressure, angiogenesis, and fibrosis in cirrhotic rats. Hepatology. 2014;60:633–47.
Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146:1339–50.
Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918–27.
Kantari-Mimoun, C.; Castells, M.; Klose, R.; Meinecke, A.K.; Lemberger, U.J.; Rautou, P.E.; Pinot-Roussel, H.; Badoual, C.; Schrödter, K.; Österreicher, C.H.; Fandrey, J.; Stockmann. C. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology 2015, 61, 2042–2055.
Calderone V, Gallego J, Fernandez-Miranda G, Garcia-Pras E, Maillo C, Berzigotti A, et al. Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology. 2016;150:982–97.
•• Bava, F.A.; Eliscovich, C.; Ferreira, P.G.; Miñana, B.; Ben-Dov, C.; Guigo, R.; Valcarcel, J.; Mendez, R. CPEB1 coordinates alternative 3′UTR formation with translational regulation. Nature 2013, 495, 121–125 Excellent work revealing a novel function of CPEB1 in mediating alternative 3′-UTR processing during cell proliferation and tumorigenesis, which is coordinated with regulation of mRNA translation, through its dual nuclear and cytoplasmic functions.
Fernandez-Miranda G, Mendez R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res Rev. 2012;11:460–72.
Pique M, Lopez JM, Foissac S, Guigo R, Mendez R. A combinatorial code for CPE-mediated translational control. Cell. 2008;132:434–48.
Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature. 2000;404:302–7.
Maillo C, Martin J, Sebastian D, Hernandez-Alvarez M, Garcia-Rocha M, Reina O, et al. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat Cell Biol. 2017;19:94–105.
Mendez R, Richter JD. Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol. 2001;2:521–9.
Garcia-Pras E, Gallego J, Coch L, Mejias M, Fernandez-Miranda G, Pardal R, et al. Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut. 2017;66:1306–20.
•• Ortiz-Zapater, E.; Pineda, D.; Martinez-Bosch, N.; Fernandez-Miranda, G.; Iglesias, M.; Alameda, F.; Moreno, M.; Eliscovich, C.; Eyras, E.; Real, F.X.; Mendez, R.; Navarro, P. Key contribution of CPEB4-mediated translational control to cancer progression. Nat Med 2011, 18, 83–90 Excellent study documenting a key role for post-transcriptional gene regulation in tumor development and describing a detailed mechanism for gene expression reprogramming underlying malignant tumor progression.
Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 increases expression of PFKFB3 to induce glycolysis and activate mouse and human hepatic stellate cells, promoting liver fibrosis. Gastroenterology 2020, S0016-5085(20)30328–0. https://doi.org/10.1053/j.gastro.2020.03.008
Funding
This work was supported by grants from the Spanish Government (SAF2017-87988-R; PRE2018-083718), the Spanish Association Against Cancer, Worldwide Cancer Research Foundation, BBVA Foundation, La Caixa Foundation, and Marato TV3 Foundation. CIBERehd is an initiative from the Instituto de Salud Carlos III. IDIBAPS is supported by the CERCA Programme (Catalan Government).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Chronic Liver Disease
Rights and permissions
About this article

Cite this article
Mejias, M., Balvey, A. & Fernandez, M. Crosstalk Between Angiogenesis and Fibrogenesis in Liver Disease. Curr. Tissue Microenviron. Rep. 1, 121–129 (2020). https://doi.org/10.1007/s43152-020-00013-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43152-020-00013-w