
Abstract
It is well established that inflammatory reactions and oxidative stress play an imperial role in cerebral ischemia-reperfusion pathogenesis. Fisetin is a flavonoid and has an antioxidant and anti-inflammatory effect on various diseases. In this study, we have been working to examine the neuroprotective effect of fisetin in brain injuries triggered by cerebral ischemic-reperfusion and explore the potential role of nuclear factor kappa B (NF-κB) signaling. In vitro, fisetin was examined against the cell viability, lactate dehydrogenase (LDH) leakage, cytokines, and apoptosis after ischemia/reperfusion (I/R) induced in the cells. In vivo, I/R injury was induced in the brain via transient middle cerebral artery occlusion (2 h) and reperfusion (20 h). The infarction area, brain water content, and neurofunctional parameters were also estimated. Inflammatory cytokines and brain injury markers were scrutinized at the end of the study. Fisetin treatment alleviated cell injury and suppressed the inflammatory cytokines (interleukin-1 (IL-1), tumor necrosis factor- α (TNF-α), inducible nitric oxide synthase (iNOS), interleukin-1β (IL-1β), cyclooxygenase-2 (COX-2), interleukin-16 (IL-6), and prostaglandin E2 (PGE2)) and antioxidant parameters in a dose-dependent manner. Fisetin significantly (P < 0.001) reduced the infarct volume, brain water content. Fisetin significantly (P < 0.001) suppressed the neurological parameters and inflammatory cytokines such as IL-1, TNF-α, iNOS, IL-1β, COX-2, IL-6, PGE2, and oxidative markers in a dose-dependent manner. Fisetin significantly (P < 0.001) reduced the inflammatory mediators including NF-κB and intercellular adhesion molecule 1 (ICAM-1). Further studies also showed that fisetin significantly inhibited the NF-κB activity via inflammatory and antioxidant pathways. In conclusion, by suppressing inflammatory cytokines, fisetin protected the brain tissue against I/R injury, and this effect could be due to reduced NF-κB activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Stroke is the second most common cause of death after coronary artery disease all over the world [1]. Stroke causes 12% death among stroke patients (50%) to die from cerebral ischemia-reperfusion [2, 3]. Ischemic stroke causes the most morbidity and mortality globally. Cerebral ischemia was induced due to blockage or reduction of blood flow into some parts of a brain region, which is induced due to organ transplantation, coronary angioplasty, contraction, percutaneous transluminal, and so on [2, 4]. Clinically, recanalization has mostly been used to restore the blood supply; also, this reperfusion may cause further brain damage and this process is also called I/R injury [2]. Thrombectomy or thrombolysis is the common therapeutics to treat recanalization. The incidence of I/R injury after stroke is often targeted at different molecular cascades further collapsing the blood-brain barrier, exacerbating brain injury [5, 6]. It also induced many insults to cerebral microvasculatures characterized by continuous generation of free radicals, endothelial cell injury, and degranulation of the mast cells. Furthermore, there are few neuroprotective treatments available which can protect the brain from ischemic brain injury [6,7,8]. Improper blood supply results in the scarcity of glucose level and oxygen supply in the different parts of the brain, directing it toward the anaerobic pathway of a cycle generating a huge amount of H+ ion and lactic acid, and their reversal to the mitochondrial matrix reducing cytoplasmic pH resulting in acidosis and formation of free radicals and inflammatory reactions [9]. PGE2, matric metalloproteinases (MMPs), cyclooxygenase-2 (COX-2), and acid-sensing ion channel (ASIC) all play a significant role in developing this condition [9]. During the induction of I/R in the brain tissue, over-production of enzymes in the tissue starts inducing oxidative stress and inflammatory reaction and finally inducing excessive neuronal death. Arachidonic acid metabolites via COX-2 and PGE2, and its over-production due to overactivation of AMPA receptor, which is the leading cause of the induction of oxidative stress and ultimately damaged the neuronal [8, 9]. Previous research suggests that inflammation and oxidative stress are the best approaches to treat the disease [6, 8, 9]. The researcher continuously tries scrutinizing the neuroprotective agent for the treatment of I/R injury in the brain. It is well known that an inflammatory reaction plays a significant role in the pathogenesis of I/R injury. Moreover, the researcher is targeting the inflammatory reaction to treat I/R injury [10, 11]. During I/R injury, shortage of blood flow results in energy failure that promotes a metabolic complex series, ultimately inducing neuronal cell death [10, 12]. The ensuing cascade of events induces a reduction in adenosine triphosphate (ATP) and mitochondrial dysfunction, which further start the generation of free radicals and boost the lipid peroxidation [10, 12]. Various published pieces of literature suggest that oxidative stress plays an imperative role in neuronal cell death after I/R injury [13, 14]. Oxidative metabolism is necessary for brain survival and is also linked with the continuous generation of ROS. During the physiologic conditions, reactive oxygen species (ROS) regulation is managed via the endogenous antioxidant defense system and maintains homeostasis [13, 15]. Any disturbance in the homeostasis resultants causes oxidative stress, which is a significant factor for the initiation of apoptosis and inflammatory reaction leading to neuronal injury. Brain tissue is susceptible to oxidative injury; moreover, it is believed that the pharmacological alteration of oxidative injury is one of the best approaches to treat cerebral ischemic-reperfusion disease [13, 16, 17].
Inflammation is the primary cause of injury to the blood-brain barrier and causes brain injury during ischemic stroke. In most cases, the inflammatory reaction is induced via necrotic or microglia cells secreting pro-inflammatory cytokines or cytotoxicity like active oxygen factors [11,12,13]. Toll-like receptors (TLRs) are the necrotic factor that secretes from necrotic cells and contributes to the promotion of inflammatory reactions. Besides, their downstream factor (p38 mitogen–activated protein kinase (MAPK)) also contributes to protection against cerebral damage caused by alteration of TLR4. NF-κB also plays an important role to boost the inflammatory reaction and the level of NF-κB boosted during I/R injury [18, 19]. The antioxidant and anti-inflammatory potential of fisetin has been observed in the previous investigation, and we also know that oxidative stress and inflammatory reaction play an important role during I/R injury.
Flavonoids, commonly found in the various plant species, are low molecular weight polyphenolic phytoconstituents and they possess various pharmacological effects such as antitumor, neuroprotective, cardioprotective, anti-inflammatory, and antioxidant effect [20, 21]. Fisetin (3,3′,4′,7-tetrahydroxyfavone) is a flavonoid, commonly present in cucumber, grapes, persimmon, strawberry, and onion. Previous study suggests that fisetin is involved in controlling the oxidative stress reaction via scavenging the free radicals and reducing the production of lipid peroxidation [20]. It also suppressed the inflammatory reaction via the reduction of cytokines and inflammatory mediators. Fisetin also exhibited chemotherapeutic agent, neuroprotective effect, and anti-inflammatory effects in rodents. Fisetin having excellent free radical scavenging activity due to its hydrophobic nature can easily enter into the cell membrane and deposit into the cells to exerts its antioxidant, neuroprotective, and neurotrophic effects [20, 21]. Fisetin showed the neuroprotective effect via reducing the inflammatory reaction and reactive gliosis during aluminum chloride (AlCl3)–induced neurotoxicity. As literature suggests, fisetin has excellent antioxidant and anti-inflammatory property against various diseases [20, 21]. Due to its antioxidant and anti-inflammatory nature, in this experimental study, we attempt to examine the neuroprotective effect of fisetin against I/R injury in rats and cell lines and to investigate the mechanism of action.
MATERIAL AND METHOD
Cell
Human neuroblastomas (SH-SY5Y) were obtained from the American Type Culture Collection, Manassas, VA, USA.
Cell Culture
PC12 cells were used for the ischemia/reperfusion (I/R) method. Dulbecco’s modified Eagle’s medium (DMEM) was used for the culture of the PC12 cells along with fetal bovine serum (10%) in the air (95%) and carbon dioxide (CO2) (5%) and the culture medium replaced every day. For induction of the I/R model, the SH-SY5Y cells were plated into culture dishes and re-culture for the next 24 h. After that, the cells were treated with fisetin for the next 24 h and finally the medium was changed with glucose-free DMEM in the hypoxia incubator (95% N2, 5% CO2) for 4 h. After induction of the ischemia, the culture medium (glucose-free DMEM) was changed and incubated again for the next 20 h (induction for reperfusion) under standard culture conditions.
Cell Viability
For the estimation of cell viability, the PC12 cells were seeded into the 96-well plates at a density of 1 × 104 cells/well and the cell was treated with different concentrations of fisetin for 24 h and finally subjected to the I/R. MTT solution (20 휇l) was mixed into each well and finally maintained for 4 h at 37 °C. The microplate reader was used for the estimation of optical density at 490 nm. The cell survival ratio was presented in the percentage via comparison with the control.
Cell Apoptosis
For the estimation of the apoptosis rate, the PC12 cells were treated with the fisetin for 24 h before subjected to the I/R. After that, the cells were washed with phosphate buffer saline (PBS) and treated with a FITC apoptosis assay kit (Sigma Aldrich, USA) following the manufacturer’s instruction. Cell apoptosis assay has been presented as percentages of total cell counts.
LDH Leakage
LDH assay kit (Institute of Biological Engineering of Nanjing Jiancheng, Nanjing, China) was used for the estimation of LDH leakage. Briefly, the culture medium was collected and cells were frayed in PBS and the cell membrane was broken to secrete the LDH in the cells and finally centrifuged the cells to get the clear cell samples. The LDH leakage was estimated from the ratio between the LDH levels in cell content and culture medium.
15-Hydroxyeicosatetraenoic Acid (15-HETE) Level
ELISA kit was used for the estimation of the 15-HETE level in the cell supernatants. Briefly, pure rats 15-HETE capture antibody. After that, the standard sample of 15-HETE was added to the plates and incubated at 37 °C for 8 hr. After the formation of the antibody-antigen enzyme-labeled antibody complex, 3,3′,5,5′-tetramethylbenzidine (TMB) (substrate) was mixed into the cell and washed with PBS and the acid was added after 15 min to stop the reaction and final absorbance was estimated at 450 nm using the enzyme standard instrument.
Cytokines
Culture and serum supernatants were collected for the determination of cytokines. Cytokines and inflammatory mediators including IL-1, COX-2, TNF-α, ICAM-1, IL-6, iNOS, IL-1β, and PGE2 were estimated using the enzyme-linked immunosorbent assay (ELISA) kits by following the manufacturer’s protocol (Institute of Biological Engineering of Nanjing Jiancheng, Nanjing, China)
In Vivo Study
Experimental Rodent
For the current experimental protocol, Sprague-Dawley (SD) rats (8–10 weeks old; 220 ± 30 g; males) were used. The SD rats were procured from the animal house and kept under polyethylene cage under standard laboratory conditions (temperature, 20 ± 5 °C; relative humidity, 70%; 12/12 h day/light cycle). In the complete experimental study, the rats received the standard food diet and tap water.
Experimental Protocol
For the induction of occlusion of the middle cerebral artery (MCAO), the rats have been divided into the following groups (Table 1). For the induction of MCAO, the rats were anesthetized using the chloral hydrate and the middle cerebral artery (MCA) was successfully separated and occluded via monofilament nylon suture for 2 h. All test and model control group rats underwent the same protocol, and the normal control rats undergo the same protocol except for arterial occlusion. The rats were anesthetized after successful induction of the reperfusion (20 h), and blood samples were collected by puncturing the retro-orbital plexus. At the end of the protocol, all group rats were euthanized and their brain tissues were successfully removed [18].
Neurological Deficits
The neurological evaluation was performed on all experimental rats after reperfusion (22 h) and before they were sacrificed by using the previously reported method with minor modification [18]. All the neurological findings were evaluated based on a scoring system (5-point scale). The neurological finding was estimated via score 0: nonneurological deficit; score 1: failure to expand forepaw; score 2: circling to the left; score 3: no motor activity; score 4: do not walk and score 5: depressed the level of consciousness [10,11,12].
Brain Water Content
After successful induction of the reperfusion (22 h), the rats were anesthetized using xylazine and ketamine and their brain tissues were removed for the estimation of brain water content [11, 22]. The olfactory bulb and cerebellum pons were successfully separated and weighed (wet weight). Drug weight was estimated after drying the tissue at 120 °C (24 h). The content of brain water was estimated using the formula below
Brain Tissue Preparation
After completing the experimental protocol, the rats were sacrificed using cervical decapitation under anesthesia condition and the brain tissues were immediately removed and homogenized in the phosphate buffer saline (pH 7) containing ethylenediaminetetraacetic acid (EDTA) to obtain the homogenate (5% w/v). The homogenate was centrifuged in the cold at 15,000 rpm at 0 °C for 15 min and the supernatant collect was to estimate various biochemical parameters [18].
Na+-K+ ATPase Activity
The previously reported method was used for the estimation of Na+-K+ activity [13]. Briefly, the ATPase activity was estimated in 2 reactions (A and B). For the reaction A, the reaction mixture contained the brain homogenate (2.0 mL) and KCl (0.2 M), MgCl2 (0.1 M), NaCl (1.0 M), and MTris-HCL buffer (0.2 M; pH 7.4). Reaction mixture B contains the brain homogenate (0.2 mL), MgCl2 (0.1 M), ouabain (10 mM), NaCl (1.0 M), and Tris-HCL buffer (0.2 M; pH 7.4). For the initiation of enzyme reaction, the 0.2-mL MATP (25 mM) was added at room temperature (37 °C) and the reaction was terminated by adding trichloroacetic acid (TCA) (1 mL) after 15 min. Finally, the mixture was centrifuged and the supernatant was separated (approximately 0.5 mL) for the estimation of inorganic phosphorous.
Antioxidant Parameters
Antioxidant parameters, viz, catalase (CAT), malonaldehyde (MDA), glutathione (GSH), glutathione peroxidase (GPx), glutathione reductase (GR), and superoxide dismutase (SOD), were estimated using the previously published protocol [12, 13, 18].
Mitochondria-Generated ROS
The ROS generated by mitochondria was estimated using the previously reported method [18].
Cytokines and Inflammatory Mediators
Cytokines including interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), and inflammatory mediators such as cyclooxygenase-2 (COX-2), nuclear kappa B factor (NF-κB), and prostaglandin E2 (PGE2) were estimated using the enzyme-linked immunosorbent assay (ELISA) kits following the manufacturer’s protocol (Institute of Biological Engineering of Nanjing Jiancheng, Nanjing, China). All cytokines and inflammatory mediators were estimated in the serum (nanograms per milliliter) and in the brain tissue (nanograms per milligram).
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 5.0 software. The data were presented as mean ± SEM in the current study. Statistical analysis was performed using one-way variance analysis (ANOVA) followed by multiple Dennett comparison tests. P < 0.05, P < 0.01, and P < 0.001 were considered to be significant, more significant, and extreme, respectively.
RESULT
Fisetin Protect the PC12 Cells from I/R-Induced Cell Apoptosis
For examination of the neuroprotective effect of fisetin on cell injury caused by I/R, PC12 cells were treated with different concentrations of fisetin for 24 h and after that were subjected to the I/R for the next 24 h. LDH leakage assay and MTT assay were used for the estimation of the neuroprotective effect of fisetin. Figure 1 shows that the cell viability was considerably reduced via I/R treatment and fisetin treatment significantly increased cell viability in a dose-dependent manner.

Fisetin treatment protected PC12 cells from I/R-induced toxicity. a Cell viability. b LDH. c Apoptosis rate. PC12 cells were treated with fisetin for 24 h and then subjected to I/R. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group.
Figure 1b shows the LDH leakage effect. I/R showed increased LDH leakage and fisetin significantly (P < 0.001) reduced the LDH leakage in a dose-dependent treatment, suggesting that fisetin has allowed PC12 cells to maintain the integrity of cells.
Similarly, the apoptosis rate was increased in I/R and dose-dependent treatment of fisetin significantly (P < 0.001) reduced the apoptosis rate (Fig. 1c).
15-HETE Content
Figure 2 shows the 15-HETE effect. I/R showed increased 15-HETE and fisetin significantly (P < 0.001) reduced the level of 15-HETE in a dose-dependent treatment, suggesting that fisetin has allowed PC12 cells to maintain the integrity of cells.

Fisetin treatment showed the effect of 15-HETE on PC12 cells from I/R injury. PC12 cells were treated with fisetin for 24 h and then subjected to I/R. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group.
Effect of Fisetin on Inflammatory Factors in PC12 Cells
To estimate the neuroprotective effect of fisetin on PC12 cells, the PC12 cells were treated with the fisetin and subjected to I/R for the next 24 h, and the level of IL-1, TNF-α, IL-6, PGE2, IL-1β, COX-2, IL-10, and NF-κB were determined using ELISA kits. Figure 3 shows the increased levels of IL-1, TNF-α, IL-6, PGE2, IL-1β, COX-2, IL-10, and NF-κB in I/R control, and fisetin treatment significantly decreased the level of IL-1, IL-1β, IL-6, IL-10, PGE2, COX-2, and NF-κB.

Fisetin treatment protected PC12 cells from I/R-induced inflammatory response. a TNF-α. b IL-1. c IL-1β. d IL-6. e IL-10. f PGE2. g COX-2. f NF-κB. PC12 cells were treated with fisetin for 24 h and then subjected to I/R. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group.
The effect of ICAM-1 on I/R induced in PC12 cells is shown in Fig. 4. The I/R group showed an increased level of ICAM-1, and dose-dependent fisetin treatment significantly (P < 0.001) reduced the level of ICAM-1.

Fisetin treatment showed the effect of ICAM-1 on PC12 cells from I/R injury. PC12 cells were treated with fisetin for 24 h and then subjected to I/R. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group.
Effect of Fisetin on Neurological Parameters
During I/R injury in the brain, the neurological score increases and a similar result was observed in I/R group rats. On day 1, the neurological score reached the maximum level and slightly reduced until day 5. Fisetin-treated group rats dose-dependently exhibited a suppressed neurological score. Nimodipine treatment resulted in a decreased neurological score (Fig. 5a).

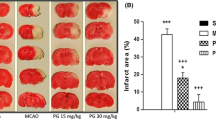
The effect of fisetin neurological parameters of I/R injury rats. a Neurological score. b Brain edema. c Infract volume. d Brain water content. e Evan blue leakage. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Figure 5 b, c, d, and e demonstrated the boosted brain edema, infarct volume, brain water content, and Evans blue leakage in I/R group rats and fisetin treatment significantly (P < 0.001) downregulated the level at a dose-dependent manner.
Effect of Fisetin on Potassium (K+), Sodium (Na+), and Na+K+ATPase Activity
Figure 6 shows the effect of fisetin on the K+, Na+, and Na+K+ATPase on I/R control group rats. I/R control group rats demonstrated increased levels of Na+ and Na+K+ATPase and decreased level of K+ compared to other groups. Fisetin-treated group rats showed a downregulated level of Na+ and Na+K+ATPase and an enhanced level of K+ compared to I/R control group rats. Nimodipine treated group rats significantly (P < 0.001) downregulated the levels of Na+ and Na+K+ATPase and enhanced the level of K+ compared to I/R control group rats.

The effect of fisetin on the Na+, K+, and Na+,K+-ATPase level in brain tissue of I/R injury rats. a Na+. b K+. c Na+,K+-ATPase. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Effect of Fisetin on Nitrate
I/R group rats showed an increased level of nitrate compared to the control group rats (Fig. 7). Fisetin significantly (P < 0.001) decreased the level of nitrate in a dose-dependent manner compared to I/R group rats. Nimodipine significantly (P < 0.001) reduced the nitrate level compared to I/R control group rats.

The effect of fisetin on the nitrate level in brain tissue of I/R injury rats. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Effect of Fisetin on ROS Level
Figure 8 shows the effect of fisetin and nimodipine on the level of ROS. I/R group rats showed a significantly (P < 0.001) boosted level of ROS, and dose-dependent fisetin treatment decreased ROS levels. Nimodipine significantly (P < 0.001) decreased the ROS level.

The effect of fisetin on the ROS level in brain tissue of I/R injury rats. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Effect of Fisetin on Carbonyl Content
Figure 9 shows the increased level of carbonyl in I/R group rats compared to the control group rats. Fisetin and nimodipine significantly (P < 0.001) suppressed the carbonyl level.

The effect of fisetin on the carbonyl content in brain tissue of I/R injury rats. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Effect of Fisetin on Antioxidant Parameters
Figure 10 shows the effect of fisetin and nimodipine on the antioxidant parameter in the serum of I/R group rats. I/R group rats showed the augmented MDA and 8-OhdG level and decreased SOD, GR, CAT, GSH, and GPx levels compared to the control group rats. Dose-dependent treatment of fisetin significantly (P < 0.001) reduced the MDA and 8-OhdG level and enhanced SOD, GR, CAT, GSH, and GPx levels compared to I/R control group rats. Nimodipine significantly (P < 0.001) upregulated the level of MDA and 8-OhdG and downregulated the level of SOD, GR, CAT, GSH, and GPx.

Showed the effect of fisetin on the antioxidant level in the serum of I/R injury rats. a SOD. b MDA. c GR. d CAT. e GSH. f GPx. g 8-OhdG. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Figure 11 shows the effect of fisetin and nimodipine on the antioxidant parameter in the brain tissue of I/R group rats. I/R group rats showed the boosted level of MDA and 8-OhdG and reduced levels of SOD, GR, CAT, GSH, and GPx, and dose-dependent treatment of fisetin significantly (P < 0.001) decreased the MDA and 8-OhdG level and increased the SOD, GR, CAT, GSH, and GPx level at a dose-dependent manner.

The effect of fisetin on the antioxidant level in the brain tissue of I/R injury rats. a SOD. b MDA. c GR. d CAT. e GSH. f GPx. g 8-OhdG. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
Effect of Fisetin on Inflammatory Mediators
Figures 12 and 13 show the effect of fisetin and nimodipine on the pro-inflammatory level in the serum and brain tissue of I/R group rats. I/R control group rats augmented IL-1, IL-1β, TNF-α, IL-6, COX-2, PGE2, and NF-κB level and reduced IL-10 levels compared to control group rats. Dose-dependent treatment of fisetin significantly (P < 0.001) reduced the levels of IL-1, IL-1β, TNF-α, IL-6, COX-2, PGE2, and NF-κB and increased the level of IL-10 compared to I/R group rats. A similar result was found in the nimodipine group rats.

The effect of fisetin on the cytokines and inflammatory level in the serum of I/R injury rats. a TNF-α. b IL-1. c IL-1β. d IL-6. e IL-10. f PGE2. g COX-2. f NF-κB. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.

The effect of fisetin on the cytokines and inflammatory level in the brain tissue of I/R injury rats. a TNF-α. b IL-1. c IL-1β. d IL-6. e IL-10. f PGE2. g COX-2. f NF-κB. Data were shown as mean ± SD. ###P < 0.001 versus control group; *P < 0.05, **P < 0.01, and ***P < 0.001 compared with I/R control group rats.
DISCUSSION
I/R injury is the most common clinical condition of circulating arrests and also induced neuronal injury in some areas of the brain such as the hippocampus [15, 18]. Various mechanisms are involved in cerebral ischemia–induced damage such as free radical injury, inflammatory response, and calcium overload [14, 15, 18]. The inflammatory response has been identified as the main cause of cerebral ischemia between them [10, 11]. The production of pro-inflammatory cytokines begins after the onset of cerebral I/R and results in injury to the tissue, improper cell repair, and dysfunction. Accordingly, targeting the inflammatory reaction during the ischemia-reperfusion injury is the best approach for effective treatment [16, 18, 19].
In the eukaryotic cells, inflammatory mediator such as NF-휅B plays a significant transcription factor which regulates the pro-inflammatory cytokines and inflammatory mediators. A significant target for reducing the inflammatory reaction was found in various diseases [3, 19]. Prior research suggests that the NF-휅B level increased during the brain injury–induced via ischemia [10, 12]. During normal conditions, NF-휅B was linked to the inhibitory protein (I휅B) and confiscated in the cytoplasm, and during the activation of NF-휅B, I휅B kinases were also activated and I휅B phosphorylation was initiated, which induced further proteasome-mediated degradation [10, 12, 18]. During the activation of NF-휅B, NF-휅B migrated to the nucleus from the cytoplasm and binds with the binding sequence to further activate the pro-inflammatory cytokines and inflammatory mediators such as TNF-α, IL-1, IL-1β, IL-6, IL-10, COX-2, and PGE2 [10, 12, 18]. This evidence indicates that NF-κB played a significant role in controlling the inflammatory reaction, and suppression of NF-κB was protective against neurodegeneration and neuroinflammation. In the current experimental study, the NF-κB level in the brain cells was increased, and fisetin significantly reduced the NF-κB level and suggested the anti-inflammatory effect.
MCAO and ischemia-reperfusion in rats were used in the current experimental study to estimate the mimics of some features of human brain pathology [18, 19]. Based on the result especially the infarct volume and neurological deficit scores, we can say that the I/R model was successfully established. Fisetin successfully reduced the infarct volume and the dose-dependent neurological deficit score. We also scrutinized the calcium-binding protein B (S-100B) and serum neuron-specific enolase (NSE) in the brain of experimental rats. Both parameters, NSE and S-100B, were used to estimate the degree of brain injury, and during I/R, the level of both parameters significantly increased and dose-dependent fisetin treatment suppressed the level. The results suggested the neuroprotective effect of fisetin against I/R-induced brain injury.
Oxidative stress and inflammatory reaction are well documented to play a significant role in I/R injury [13, 18, 19]. I/R is involved in the induction of apoptosis and the accumulation of ROS. Prior research suggests the production of free radicals commonly observed in I/R injury and finally causes oxidative stress [13, 18, 19]. During I/R, the cascade reactions start such as excitotoxicity, oxidative stress, and inflammatory reaction and finally induced the neuronal cell death [10, 12]. Endogenous antioxidant enzymes including GSH, CAT, GPx, and SOD play an important role to reduce oxidative stress and play a protective role in neuronal damage against reactive oxygen species–induced cell apoptosis [10, 11]. MDA is a lipid peroxidation parameter and plays an important role to inhibit lipid peroxidation in the tissue. SOD is the primary line of endogenous antioxidant enzymes and plays a significant role to protect tissue against ROS damage. SOD catalyzes the O2 (superoxide anion) transferred to H2O2 (hydrogen peroxide) and finally reduced the OH (hydrogen radical) generation. CAT also catalyzes the O2 transferred to H2O2 along with SOD and finally reduced the OH radical generation [10, 11, 23]. As for other endogenous antioxidant enzymes, GSH directly reacts with the ROS and plays a co-factor with an enzyme and GPx reduces the H2O2 and lipid peroxide levels in tissue [10]. In the current experimental study, the I/R group showed increased levels of MDA and reduced level of SOD, GSH, CAT, and GPx suggesting the oxidative stress induced in rats suffering from I/R. Fisetin significantly (P < 0.001) reduced MDA levels and increased SOD, GSH, GPx, and CAT level and suggested the antioxidant potential.
Due to the increased level of cytokines into the brain tissue and circulation, increased leukocytes into the tissue and circulation, which directly showed the effect to the accumulation of neutrophils, activated microglia and macrophages, which invade the ischemic region and cause loss of neuronal cells and brain tissue, can increase the area of the cerebral infarction [24, 25]. I/R-induced rats showed an increased level of infarct damage, and dose-dependent treatment of fisetin significantly reduced the infarct damage and suggested the brain tissue–protecting effects. Prior research suggests that the brain swelling commonly found during ischemic stroke is due to the induction of blood reperfusion [23, 25]. Except for the portion of the fluid, a defined tissue mass and new mass tissue increase in the extracellular region of the brain and cause brain swelling [17, 19]. The researcher targeted the cerebral infarct area and brain swelling in the brain tissue to estimate the potential of drugs tested. Brain tissue swelling was determined by the ratio of the left and right area and brain weight, instead of calculating the wet and dry brain weight [13, 16]. Yuan et al. estimated the cerebral infarct area and brain swelling in the brain tissue and suggesting the neuroprotective effect of baicalein against the ischemic/reperfusion injury [26]. I/R-induced rats showed increased infarct area and cerebral swelling in the brain area, and fisetin considerably reduced the infarct area and cerebral swelling. Based on the outcome, we can conclude that the fisetin has the potential neuroprotective effect. Clinically, ischemic cerebral edema became a prevalent factor in global I/R injury.
Na+,K+-ATPase is strongly localized in the neuronal cell membrane and usually retains Na+ and K+ concentrations intracellular [13, 27]. During the ischemia, the level of Na+,K+-ATPase in the brain tissue was considerably reduced due to decreased ATP content and enhanced production of the enzymatic inhibitor, which take part in ionic disorder [28]. Na+,K+-ATPase dysfunction in the cell membrane triggered depolarization of the membrane and aggregation of Na+ and water in the cell, which eventually mediated cytotoxic edema in the ischemic cell [13]. In this experimental study, fisetin considerably reduced Na+ and K+ and finally reduced the activity of Na+,K+-ATPase by inducing a lower net ion shift, which resulted in less edema formation. During the cerebral ischemic reperfusion injury, the levels of pro-inflammatory cytokines and inflammatory mediators were considerably boosted due to some pathological stimulus [13, 29].
It is well documented that early inflammation during I/R injury is classified via the recruitment of vascular macrophages and endogenous microglia. Previous studies suggest that the inflammatory cells exhibited the production of pleiotropic mediators such as cytokines, chemokines, and prostanoids [9]. The alteration in the production of numerous anti- and pro-inflammatory includes chemokines in the brain tissue that are the part of the inflammatory and immune response following stroke [9]. Linag et al. showed the neuroprotective effect of umbelliferone against the cerebral ischemia/reperfusion via suppression of COX-2 and PGE2 against the cerebral ischemia/reperfusion in rats [30]. Previous research has shown that COX-2 and PGE2 contribute to brain injury through the generation of reactive oxygen species and prostanoids, while iNOS, which produces a large amount of nitric oxide, initiates oxidative stress and DNA damage [11, 18, 29]. The cytokines and inflammatory mediators are secreted via immune cells and also generated via brain cells such as neurons and glial cells [16, 19]. The most effective cytokines and inflammatory mediators linked with brain inflammation during ischemia are IL-1, IL-1β, PGE2, IL-6, COX-2, IL-10, TNF-α, and NF-κB. Among all cytokines and inflammatory mediators, TNF-α and IL-1 could exacerbate the degree of brain injury. Pro-inflammatory cytokines such as TNF-α interrupt the blood-brain barrier (BBB) and therefore encouraged the infiltration of systemic inflammatory cells into the damaged brain and start the secretion into the circulation. This reaction boosted the local inflammatory reaction after cerebral ischemia [13,14,15]. Peroxisome proliferator–activated receptor gamma (PPAR-γ) agonists are commonly known to reduce the level of various inflammatory mediators and suppressing the post-ischemic expression of key inflammatory genes such as COX-2, PGE2, and TNF-α in brain tissue. PPAR-γ antagonist activates NF-κB and signal transducer and is an activator of transcription factors and transcription-1 (STAT1), which play an important role in the alteration of inflammatory mediators after cerebral ischemia. In the current experimental study, I/R showed an increased level of COX-2, PGE2, TNF-α, and NF- κB and dose-dependent treatment of fisetin considerably reduced their level and suggested the anti-inflammatory effect. Prior reports suggest that the inflammatory reaction contributes to stroke-induced morbidities, which can be directly or indirectly involved in ischemic injury and contribute to serious side effects such as damaging the brain tissue [9,10,11]. NF-κB (a transcription factor) plays a significant role in the regulation of numerous inflammatory genes such as COX-2, IL-6, TNF-α, and PGE2. The NF-κB is activated during I/R injury and activated the inflammatory reaction as well as oxidative stress, which contributed to neuronal death and deoxyribonucleic acid (DNA) damage [9,10,11, 15, 17]. Both damages further expand the damage into the brain tissue and cause strokes. We found in the current experimental study that cytokines and inflammatory mediators IL-1, PGE2, IL-1β, IL-6, TNF-α, COX-2, IL-10, and NF-κB significantly increased after the I/R and dose-dependent treatment of fisetin significantly reduced the level of IL-1, PGE2, IL-1β, IL-6, TNF-α, COX-2, IL-10, and NF-κB in PC12 cells, brain tissue, and experimental rat serum [18, 19]. Yang et al. showed the neuroprotective effect of tangeretin against the cerebral ischemia-reperfusion injury in rats via inflammatory pathway [31]. A similar result was observed in the current experimental study. In this experimental study, we have observed that fisetin is attributable to the suppression of the inflammatory reaction as a result of the downregulation of COX-2, PGE2, TNF-α, IL-1, NF-κB, IL-1β, and IL-10 as a mediator of the activation of PPAR-γ.
CONCLUSION
Our results clearly suggest that fisetin exhibits a neuroprotective effect against I/R-induced inflammation and oxidative stress in in vitro and in vivo models. Fisetin treatment protected the cells from I/R injury inflammation via the reduction of NF-κB-mediated inflammatory response. Fisetin significantly reduced the neurological defect, infarct volume, and brain edema. Fisetin exhibited a neuroprotective effect against I/R-induced cerebral ischemic-reperfusion injury via alteration of oxidative and inflammatory mediators. The above finding specifically describes the molecular process important to the neuroprotective activity of fisetin and may offer valuable insight into a safer design of neuroprotective agents against ischemic stroke. These findings provided some scientific evidence for the cerebral protective effect of fisetin and suggested that it is beneficial for treating various inflammatory-related brain diseases.
Abbreviations
- NF-κB:
-
nuclear factor kappa B
- LDH:
-
lactate dehydrogenase
- I/R:
-
ischemia/reperfusion
- IL-1:
-
interleukin-1
- TNF-α:
-
tumor necrosis factor-α
- Inos:
-
inducible nitric oxide synthase
- IL-1β:
-
interleukin-1β
- COX-2:
-
cyclooxygenase-2
- IL-6:
-
interleukin-6
- PGE2 :
-
prostaglandin E2
- MMPs:
-
matric metalloproteinases
- ASIC:
-
acid-sensing ion channel
- ATP:
-
adenosine triphosphate
- ROS:
-
reactive oxygen species
- TLRs:
-
toll-like receptors
- MAPK:
-
mitogen-activated protein kinase
- AlCl3 :
-
aluminum chloride
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- CO2 :
-
carbon dioxide
- PBS:
-
phosphate buffer saline
- 15-HETE:
-
15-hydroxyeicosatetraenoic acid
- TMB:
-
3,3′,5,5′-tetramethylbenzidine
- SD:
-
Sprague-Dawley
- MCAO:
-
middle cerebral artery
- EDTA:
-
ethylenediaminetetraacetic acid
- CAT:
-
catalase
- MDA:
-
malonaldehyde
- GSH:
-
glutathione
- GPx:
-
glutathione peroxidase
- GR:
-
glutathione reductase
- SOD:
-
superoxide dismutase
- K+ :
-
potassium
- Na+ :
-
sodium
- PPAR-γ:
-
peroxisome proliferator–activated receptor gamma
- STAT1:
-
signal transducer and activator of transcription factors and transcription-1
- DNA:
-
deoxyribonucleic acid
References
Song, Jungbin, Young Sik Kim, Dong Hwan Lee, Sung Hyun Lee, Hyo Jin Park, Donghun Lee, and Hocheol Kim. 2019. Neuroprotective effects of oleic acid in rodent models of cerebral ischaemia. Scientific Reports 9: 10732. https://doi.org/10.1038/s41598-019-47057-z.
Huang, Lifa, Chengwei Chen, Xin Zhang, Li Xu, Zupeng Chen, Chao Yang, Xiaolong Liang, Guochong Zhu, and Xu. Zhen. 2018. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. Journal of Molecular Neuroscience 64: 129–139. https://doi.org/10.1007/s12031-017-1006-x.
Fu, Chen, Xinyang Zhang, Zixiu Zeng, Tian Yang, Xianglan Jin, Fengli Wang, Zhenmin Xu, Baoxin Chen, Hong Zheng, and Xuemei Liu. 2020. Neuroprotective effects of Qingnao dripping pills against cerebral ischemia via inhibiting NLRP3 Inflammasome signaling pathway: in vivo and in vitro. Frontiers in Pharmacology 11: 65. https://doi.org/10.3389/fphar.2020.00065.
Chen, Chunxia, Chen Wan, Zhihuan Nong, Yichu Nie, Xiaoyu Chen, Xiaorong Pan, Ying Guo, Meicun Yao, and Wenbin Deng. 2020. Hyperbaric oxygen alleviated cognitive impairments in mice induced by repeated cerebral ischemia-reperfusion injury via inhibition of autophagy. Life Sciences 241: 117170. https://doi.org/10.1016/j.lfs.2019.117170.
Li, Kang, Dun Ding, and Ming Zhang. 2016. Neuroprotection of osthole against cerebral ischemia/reperfusion injury through an anti-apoptotic pathway in rats. Biological and Pharmaceutical Bulletin 39: 336–342. https://doi.org/10.1248/bpb.b15-00699.
Guo, Minmin, Huiling Lu, Jian Qin, Qu Shengbiao, Wenbo Wang, Yanhong Guo, Weiyong Liao, Mengwei Song, Jian Chen, and Yong Wang. 2019. Biochanin A provides neuroprotection against cerebral ischemia/reperfusion injury by Nrf2-mediated inhibition of oxidative stress and inflammation signaling pathway in rats. Medical Science Monitor 25: 8975–8983. https://doi.org/10.12659/MSM.918665.
Gao, Jianmei, Nana Chen, Na Li, Fan Xu, Wei Wang, Yaying Lei, Jingshan Shi, and Qihai Gong. 2020. Neuroprotective effects of trilobatin, a novel naturally occurring Sirt3 agonist from Lithocarpus polystachyus Rehd., mitigate cerebral ischemia/reperfusion injury: involvement of TLR4/NF-κB and Nrf2/Keap-1 signaling. Antioxidants and Redox Signaling 33: 117–143. https://doi.org/10.1089/ars.2019.7825.
Dai, Yunyi, Haojie Zhang, Jianping Zhang, and Mingguang Yan. 2018. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-κB pathway. Chemico-Biological Interactions 284: 32–40. https://doi.org/10.1016/j.cbi.2018.02.017.
Maurya, Khushboo, and AnandKumar Pandey. 2019. Molecular docking study for evaluation of neuroprotective potential of sericin against cerebral stroke and exploring its biomaterial properties. Biomedical Research Journal 6: 17. https://doi.org/10.4103/bmrj.bmrj_5_19.
Yaidikar, Lavanya, Bavya Byna, and Santh Rani Thakur. 2014. Neuroprotective effect of punicalagin against cerebral ischemia reperfusion-induced oxidative brain injury in rats. Journal of Stroke and Cerebrovascular Diseases 23: 2869–2878. https://doi.org/10.1016/j.jstrokecerebrovasdis.2014.07.020.
Sinha, Kusum, Geeta Chaudhary, and Yogendra Kumar Gupta. 2002. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sciences 71: 655–665. https://doi.org/10.1016/S0024-3205(02)01691-0.
Jiang, Jun, Wei Wang, Yong Jun Sun, Mei Hu, Fei Li, and Dong Ya Zhu. 2007. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. European Journal of Pharmacology 561: 54–62. https://doi.org/10.1016/j.ejphar.2006.12.028.
Tang, Hao, Yuping Tang, Nianguang Li, Qianping Shi, Jianming Guo, Erxin Shang, and Jin Ao Duan. 2014. Neuroprotective effects of scutellarin and scutellarein on repeatedly cerebral ischemia-reperfusion in rats. Pharmacology Biochemistry and Behavior 118: 51–59. https://doi.org/10.1016/j.pbb.2014.01.003.
Qian, Lihua, Minzhe Shen, Hao Tang, Yuping Tang, Li Zhang, Fu Yifan, Qianping Shi, and Nian Guang Li. 2012. Synthesis and protective effect of scutellarein on focal cerebral ischemia/reperfusion in rats. Molecules 17: 10667–10674. https://doi.org/10.3390/molecules170910667.
Sun, Lingyan, Xia Tian, Lingshan Gou, Xin Ling, Ling Wang, Yan Feng, Xiaoxing Yin, and Yi Liu. 2013. Beneficial synergistic effects of concurrent treatment with theanine and caffeine against cerebral ischemia-reperfusion injury in rats. Canadian Journal of Physiology and Pharmacology 91: 562–569. https://doi.org/10.1139/cjpp-2012-0309.
Tang, Hao, Ze Xi Dong, Gu Ting, Nian Guang Li, Yu Ping Tang, Qian Ping Shi, Jian Ming Guo, Peng Xuan Zhang, and Jin Ao Duan. 2015. Studies on the protective effects of scutellarein against neuronal injury by ischemia through the analysis of endogenous amino acids and Ca2+ concentration together with Ca2+-ATPase activity. Journal of Chemistry 2015: 497842–497847. https://doi.org/10.1155/2015/497842.
Wang, Wenjuan, Xiaotang Ma, Jichun Han, Mingjie Zhou, Huanhuan Ren, Qunwen Pan, Chunli Zheng, and Qiusheng Zheng. 2016. Neuroprotective effect of scutellarin on ischemic cerebral injury by down-regulating the expression of angiotensin-converting enzyme and AT1 receptor. PLoS ONE 11 (1): e0146197. https://doi.org/10.1371/journal.pone.0146197.
Du, Shibin, Youliang Deng, Hongjie Yuan, and Yanyan Sun. 2019. Safflower Yellow B Protects brain against cerebral ischemia reperfusion injury through AMPK/NF-kB pathway. Evidence-based Complementary and Alternative Medicine 2019: 7219740–7219711. https://doi.org/10.1155/2019/7219740.
Liu, Xiao Jie, Zhi Gang Mei, Jing Ping Qian, Yong Bao Zeng, and Ming Zhi Wang. 2013. Puerarin partly counteracts the inflammatory response after cerebral ischemia/reperfusion via activating the cholinergic anti-inflammatory pathway. Neural Regeneration Research 8: 3203–3215. https://doi.org/10.3969/j.issn.1673-5374.2013.34.004.
Akpa, Amaka Rosita, Joseph Olusegun Ayo, Hudu Garba Mika’il, and Friday Ocheja Zakari. 2020. Protective effect of fisetin against subchronic chlorpyrifos-induced toxicity on oxidative stress biomarkers and neurobehavioral parameters in adult male albino mice. Toxicological Research. Springer Singapore. https://doi.org/10.1007/s43188-020-00049-y.
Piao, Mei Jing, Ki Cheon Kim, Sungwook Chae, Young Sam Keum, Hye Sun Kim, and Jin Won Hyun. 2013. Protective effect of fisetin (3,7,3’,4’-tetrahydroxyflavone) against γ-irradiation-induced oxidative stress and cell damage. Biomolecules and Therapeutics 21: 210–215. https://doi.org/10.4062/biomolther.2013.017.
Shin, Won Ho, Sang Joon Park, and Eun Joo Kim. 2006. Protective effect of anthocyanins in middle cerebral artery occlusion and reperfusion model of cerebral ischemia in rats. Life Sciences 79: 130–137. https://doi.org/10.1016/j.lfs.2005.12.033.
Bora, Kundan Singh, and Anupam Sharma. 2010. Neuroprotective effect of Artemisia absinthium L. on focal ischemia and reperfusion-induced cerebral injury. Journal of Ethnopharmacology 129: 403–409. https://doi.org/10.1016/j.jep.2010.04.030.
Huang, Judy, Urvashi M. Upadhyay, and Rafael J. Tamargo. 2006. Inflammation in stroke and focal cerebral ischemia. Surgical Neurology 66: 232–245. https://doi.org/10.1016/j.surneu.2005.12.028.
Pan, Jie, Angelos Aristeidis Konstas, Brian Bateman, Girolamo A. Ortolano, and John Pile-Spellman. 2007. Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology. 49 (2): 93–102. https://doi.org/10.1007/s00234-006-0183-z.
Yuan, Yu, Weidong Men, Xiaosong Shan, Hexin Zhai, Xiaoxia Qiao, Lianting Geng, and Chunhui Li. 2020. Baicalein exerts neuroprotective effect against ischaemic/reperfusion injury via alteration of NF-kB and LOX and AMPK/Nrf2 pathway. Inflammopharmacology 28: 1327–1341. https://doi.org/10.1007/s10787-020-00714-6.
Hu, Xia Min, Mi Mei Zhou, Xian Min Hu, and Fan Dian Zeng. 2005. Neuroprotective effects of scutellarin on rat neuronal damage induced by cerebral ischemia/reperfusion. Acta Pharmacologica Sinica 26: 1454–1459. https://doi.org/10.1111/j.1745-7254.2005.00239.x.
Martz, Dean, Mary Beer, and A. Lorris Betz. 1990. Dimethylthiourea reduces ischemie brain edema without affecting cerebral blood flow. Journal of Cerebral Blood Flow and Metabolism 10: 352–357. https://doi.org/10.1038/jcbfm.1990.64.
Tang, Hao, Yuping Tang, Nian Guang Li, Hang Lin, Weixia Li, Qianping Shi, Wei Zhang, Pengxuan Zhang, Zexi Dong, Minzhe Shen, Ting Gu, and Jin-Ao Duan. 2015. Comparative metabolomic analysis of the neuroprotective effects of scutellarin and scutellarein against ischemic insult. PLoS ONE 10 (7): e0131569. https://doi.org/10.1371/journal.pone.0131569.
Liang, Sen, Zhaoyao Chen, Hui Li, Zhilan Cang, Kailin Yin, Minghua Wu, and Shouzhen Luo. 2020. Neuroprotective effect of umbelliferone against cerebral ischemia/reperfusion induced neurological deficits: in-vivo and in-silico studies. Journal of Biomolecular Structure and Dynamics 0. Taylor & Francis: 1–11. https://doi.org/10.1080/07391102.2020.1780153.
Yang, Tiansong, Chuwen Feng, Dongyan Wang, Yuanyuan Qu, Yan Yang, Yulin Wang, and Zhongren Sun. 2020. Neuroprotective and anti-inflammatory effect of tangeretin against cerebral ischemia-reperfusion injury in rats. Inflammation 43. Inflammation: 2332–2343. https://doi.org/10.1007/s10753-020-01303-z.
Acknowledgments
The authors are very thankful to the Xi’an No.1 Hospital for providing the necessary facility.
Data and Materials Availability
All the data are provided on the request to the corresponding author.
Author information
Authors and Affiliations
Contributions
P. Z. performed the experimental study. J. C. designed the experimental study and interpreted the data. Both authors equally contributed to the proofreading.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
The whole animal study was approved from Institute.
Consent for Publication
N/A
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, P., Cui, J. Neuroprotective Effect of Fisetin Against the Cerebral Ischemia-Reperfusion Damage via Suppression of Oxidative Stress and Inflammatory Parameters. Inflammation 44, 1490–1506 (2021). https://doi.org/10.1007/s10753-021-01434-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-021-01434-x