
Abstract
Salinity tolerance in rice is critical at reproductive stage because it ultimately determines grain yield. An F2 mapping population derived from a Sadri/FL478 cross was exposed to saline field conditions (6–8 dS m − 1) after the active tillering stage to identify reproductive stage specific QTLs for salinity tolerance. Genetic linkage map was constructed using 123 microsatellite markers on 232 F2 progenies. Totally 35 QTLs for 11 traits under salinity stress were detected with LOD > 3, out of which 28 QTLs that explained from 5.9 to 30.0% phenotypic variation were found to be significant based on permutation test. Three major QTL clusters were found on chromosomes 2 (RM423–RM174), 4 (RM551–RM518) and 6 (RM20224–RM528) for multiple traits under salinity stress. Both parental lines contributed additively for QTLs identified for the yield components. A majority of the QTLs detected in our study are reported for the first time for reproductive stage salinity stress. Fine-mapping of selected putative QTLs will be the next step to facilitate marker-assisted backcrossing and to detect useful genes for salinity tolerance at the reproductive stage in rice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salinity is one of the major environmental stress that limit the productivity of rice (Oryza sativa L.) because of the crop’s sensitivity to high concentrations of salt in the soil (Flowers and Yeo 1995). A soil is considered saline if the electrical conductivity of its saturated extract (ECe, average root-zone EC of saturated soil extract) is above 4 dS m − 1 (United States Salinity Laboratory Staff 1954). More than 90% of world’s rice production comes from Asia, especially south and southeast Asia that has about 21.5 million ha as salt affected, of which 12 million ha are saline and 9.5 million ha are alkaline/sodic (Munns and Tester 2008). Over half of the world’s population depends on rice as a staple food crop (Narciso and Hossain 2002). Rice is relatively salt sensitive, and it has a threshold salinity of 3 dS m − 1, above which yield loss occurs (Maas and Hoffmann 1977). Maas and Grattan (1999) indicated that rice yield decrease 12% for every unit (dS m − 1) increase in ECe above 3 dS m − 1. Rice is relatively tolerant of salt stress during germination, active tillering and toward maturity, and is sensitive during the early seedling and reproductive stages (panicle initiation, anthesis and fertilization) (Zheng et al. 2001; Singh et al. 2007). Symptoms of salinity stress include white tips of affected leaves, plant stunting, reduced tillering, patchy field growth and in severe cases, plant death. Salinity significantly reduces tiller number per plant, spikelet number per panicle, fertility, panicle length and primary branches per panicle (Heenan et al. 1988; Cui et al. 1995; Khatun et al. 1995; Zeng et al. 2002; Sarhadi et al. 2012). Reduction in tiller number per plant and spikelet number per panicle were reported to be the major causes of yield loss in one cultivar of rice under salinity stress (Zeng and Shannon 2000). The number of spikelets per panicle was determined to be the most sensitive yield component. This component is determined at the early reproductive stage, around panicle initiation (PI) (Counce et al. 2000). Rao et al. (2008) reported a grain yield reduction by 27, 46 and 50% at an ECe of 8 dS m − 1 in tolerant, moderately tolerant and susceptible rice cultivars, respectively. Considerable variation for yield components was reported based on the evaluation of several diverse genotypes for salinity stress response (Yeo and Flowers 1982; Zeng et al. 2002; Moradi et al. 2003).
QTLs for salinity tolerance
The vast genetic variability reported in rice in response to salinity makes it amenable to genetic manipulation to further enhance its tolerance (Akbar et al. 1972; Flowers and Yeo 1981). Breeders have long made use of the high salinity tolerance in landraces such as Nona Bokra and Pokkali. However, negative characters in traditional varieties and numerous complex traits involved in salinity tolerance have presented challenges for conventional breeding to make significant progress and have led to increased interest in molecular breeding methods (Gregorio et al. 2002; Ismail et al. 2007; Thomson et al. 2010). QTL mapping enables the dissection of the genetic control of each tolerance mechanism, opening up the possibility of future efforts to develop varieties with improved salinity tolerance by precisely transferring QTLs into popular varieties and pyramiding multiple relevant QTLs for a particular stress-prone environment (Thomson et al. 2010).
Most previous studies identified QTLs associated with seedling stage salinity tolerance in rice. Salt tolerance has often been found associated with a lower accumulation of sodium (Na + ) in the shoot, but not always (Genc et al. 2007). Using Na + accumulation and other measures of salt tolerance, major and minor QTLs have been mapped in various crop species including rice (Flowers and Flowers 2005; Jenks et al. 2007). A number of mapping studies have identified QTLs associated with salinity tolerance in rice (Singh et al. 2007; Haq et al. 2010; Singh and Flowers 2010). A major QTL located on chromosome 1 was identified for salt tolerance using F8 recombinant inbred lines (RILs) of an IR29/Pokkali cross (Gregorio 1997). This major QTL designated Saltol, governed Na–K uptake ratio and accounted for 64.3% of the phenotypic variation in salt tolerance. SSR markers RM8094 and RM10745 were suggested for use in marker-assisted selection (MAS) of the Saltol QTL (Mohammadi-Nejad et al. 2008). Further, an analysis of single feature polymorphisms in the Saltol region suggested that FL478 contained >1 Mb DNA fragment from Pokkali at 10.6–11.5 Mb on chromosome 1, flanked by IR29 alleles (Kim et al. 2009). A study employing an F2:3 population derived from the tolerant indica landrace Nona Bokra and the susceptible japonica Koshihikari identified several QTLs controlling tolerance traits, including major QTLs for shoot K + concentration on chromosome 1 (qSKC-1) and shoot Na + concentration on chromosome 7 (qSNC-7; Lin et al. 2004). The SKC1 gene, which lies within the Saltol region, was subsequently cloned and found to encode a sodium transporter that helps to control Na + and K + homeostasis under salt stress (Ren et al. 2005). Ammar et al. (2009) used an F2:3 mapping population derived from an indica/indica cross of CSR27 (tolerant) and MI-48 (sensitive) to identify QTLs for Na + , K + and Cl − concentrations in the leaf tissue at the reproductive stage. They detected QTLs for Cl − , Na–K ratio and Na + in the leaf at the reproductive stage on chromosomes 2, 3 and 8, respectively. The three QTLs explained phenotypic variance in the range of 26–52%. Salinity tolerance at the seedling and reproductive stages is only weakly associated (Mishra B., Akbar M. and Seshu D. V. 1990 Genetic studies on salinity tolerance in rice towards better productivity in salt affected soils. Paper presented at the Rice Research Seminar, 12 July 1990, IRRI, Los Baños, Philippines; Moradi et al. 2003; Singh and Flowers 2010), suggesting that tolerance at these two stages is regulated by different sets of genes. The reproductive stage is crucial as it ultimately determines grain yield; however, the importance of the seedling stage cannot be ignored as it affects crop establishment. Hence, pyramiding of contributing traits/QTLs conferring tolerance at both stages is needed for developing resilient salt-tolerant cultivars (Moradi et al. 2003). The major objectives of this study were to identify and map the major QTLs associated with reproductive stage salinity tolerance in rice.
Materials and methods
Plant materials and population development
The F2 population from the Sadri/FL478 cross (IR96469) was used for phenotyping under salinity stress at the reproductive stage and the identification of associated QTL regions. The parents were selected based on their performance in a diallel cross analysis under salinity stress (unpublished data). Sadri (IRGC acc. 32329) is an aromatic landrace from northern Iran, susceptible to salinity at the seedling stage but tolerant at the reproductive stage. Sadri is a general name for premium-quality rice landraces from Iran. Before 2001, Sadri landraces were cultivated on more than 60% of the total rice area, because farmers refused to adopt improved varieties on account of their poor cooking quality. FL478 (IR66946-3R-178-1-1) is a recombinant-inbred line of an IR29/Pokkali cross with high seedling stage salinity tolerance, but it is sensitive at the reproductive stage. A total of 232 F2 plants were developed from the Sadri/FL478 cross and used for phenotyping as well as genotyping.
Phenotyping for yield components under salt stress
The phenotyping was conducted at International Rice Research Institute, Phillipines, under artificially salinized concrete blocks (8 × 4.8 m and 0.5 m depth). The 21-day-old seedlings of parents and 232 F2 progenies were transplanted in saline plots. Saline water solution was prepared by dissolving technical grade salt (NaCl) in water to salinize the soil by irrigation of saline water. Salinity stress was imposed initially by irrigating the saline water of ECe 3–4 dS m − 1 during transplanting at the seedling stage, which increased to ECe 6–8 dS m − 1 after the active tillering stage until maturity. The ECe of the soil was monitored and adjusted whenever necessary by saline water irrigation. At maturity, data were recorded on each F2 plant for the following characters: number of days to flowering (DFL), plant height (PHt in cm), number of panicles per plant (PnN), panicle length (PnL in cm), straw dry weight per plant (StDW in g), number of fertile spikelets per plant (FrSp), number of sterile spikelets per plant (StSp), number of total spikelets per plant (TSp), grain yield per plant (GYld in g), percent spikelet fertility (SpFr) and 1000-grain weight (TGW in g).
Construction of a linkage map and QTL mapping
Three-to-four weeks after transplanting, young leaves from all 232 F2 plants and parents were collected. Genomic DNA samples were extracted by using a modified CTAB protocol (Thomson et al. 2006). A parental survey was performed using 450 rice SSR markers. PCR amplification was performed in a 15 μL volume containing 2 μL of 25 ng genomic DNA, 1.5 μL of 10× PCR buffer (containing 100 mM Tris-HCl, pH 8.3, 500 mM KCl and 15 mM MgCl2), 1.25 μL of 1 mM dNTP, 0.5 μL of 50 mM MgCl2, 1 μL each of 5 μM forward and reverse primers and 1 μL of 5 U/μL Taq DNA polymerase with 6.75 μL sterile nano-pure H2O. These items were dispensed in each well along with 1 drop of mineral oil and then covered with PCR plate sealing film. PCR was performed in a G-Storm Thermal Cycler (Model GS1, Gene Technologies, Essex, UK) by initial denaturation at 94°C for 5 min, and then 35 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 45 s, extension at 72°C for 1 min and 30 s and final extension at 72°C for 8 min and storage at 10°C.
PCR was performed on 96-well plates, and the DNA fragments were separated on 8% acrylamide gels (C.B.S. Scientific, Del Mar, USA), and stained with SYBR-Safe (Invitrogen, Carlsbad, USA) for manual allele scoring. Polymorphic markers were used to genotype the F2 population and to construct a linkage map to cover the entire genome. The linkage map was constructed using Map Manager QTX, vQTXb20 (Manly et al. 2001). The phenotypic and genotypic data were analysed by composite interval mapping (CIM) using QGene ver. 4.3.8 (Joehanes and Nelson 2008). Permutations of 10000 iterations were used to determine the threshold of the QTLs in Qgene. Subsequently, the LOD values at P < 0.05 were used as the threshold to declare the significance of the QTLs. QTL names were designated following the standard rice QTL nomenclature (McCouch and CGSNL 2008).
Results
Phenotyping of the mapping population
The analysis of variation showed a significantly different response among the F2 progenies for the 11 evaluated traits under salt stress at the reproductive stage. Considerable effect due to salinity was observed for most of the traits evaluated at that stage.
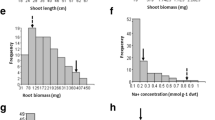
For all the traits studied, F2 population variation was beyond the range of their parents, suggesting transgressive segregation and this was observed in both directions for all the traits but to different extents. The distribution within each trait also indicated that these traits are under polygenic control and both parents, Sadri and FL478, contributed genes for these traits. The frequency distribution of yield components of the F2 population did not fit the normal distribution, except for the distribution of DFL, PHt and PnL, which were close to normal distribution (figure 1). Deviation from the normal distribution could be the result of the influence of some major genes. The phenotypic distributions of all examined traits for the F2 population displayed a continuous segregation with the skewness ranging from −0.48 to 1.34 indicating quantitative inheritance (table 1). The mean distributions for yield components showed a moderate to high transgressive segregation with values either larger or smaller than those of the parents (figure 1). The mean values of StDW, SrSp and GYld were skewed towards the lower values, while SpFr, TGW and PHt were skewed towards the higher values. Zang et al. (2008) reported that the introgression lines (BC2F8) derived from an indica/japonica cross (IR64 and Binam are both moderately tolerant of salinity) showed great variation in salinity tolerance.

Frequency distribution of yield components of Sadri/FL478 F2 progenies grown under salt stress at the reproductive stage in artificially salinized field conditions (S, Sadri; F, FL478).
Correlation among grain yield per plant and yield components
Correlation analysis can provide an understanding of the relationship of grain yield per plant (GYld) and yield components under salinity stress. Correlation analysis revealed that plant height (0.37**), panicle length (0.35**), number of panicles (0.36**), straw dry weight (0.40**), number of fertile spikelets (0.97**), spikelet fertility (0.64**) and 1000-grain weight (0.44**) had a positive and significant contribution to GYld (table 2). Number of days to flowering had a negative and weak correlation with GYld (−0.15*). Number of sterile spikelets had no significant correlation with GYld (here ‘*’ and ‘**’ indicates significance at P ≤ 0.05 and P ≤ 0.01, respectively).
Construction of a linkage map and QTL identification
Of the 450 SSR markers used for the parental survey, 155 (34.4%) showed polymorphism between the two parents. These polymorphic markers were then applied in 232 unselected F2 samples. Out of 155 polymorphic markers, 123 exhibited clear bands. The amplified fragments ranged in size from 83 to 299 bp. Linkage maps of 12 chromosomes were created based on genotypic data of 232 F2’s from Sadri/FL478, with 123 SSR primers using the Kosambi mapping function (Kosambi 1944) of Map Manager QTX, vQTXb20 (Manly et al. 2001). Marker ordering was based on published microsatellite marker information (Temnykh et al. 2001). The total size of the linkage map was 1852 cM, with an average of 15 cM, and the distance between markers ranged from the smallest interval of 0.9 cM on chromosome 3 to the largest interval of 60.6 cM on chromosome 4 (figure 2).

Genetic linkage map showing the location of QTLs for yield components under salinity stress (EC = 6–8 dS m − 1) in the Sadri/FL478 F2 population. Distances are in Kosambi centimorgans. The legends on the chromosomes represent putative regions of QTLs for each trait.
QTLs associated with yield components under salinity stress were identified through interval mapping (IM) and composite interval mapping (CIM; Zeng 1993, 1994) using QGene, ver. 4.3.8 (Joehanes and Nelson 2008). A total of 35 QTLs for 11 yield and yield-related traits were detected on all 12 chromosomes of rice except chromosomes 11 and 12 (table 3). The data were also analysed using CIM by Windows QTL Cartographer 2.5.009 (Wang et al. 2011). The results of the QTL analysis from QTL Cartographer were similar to the QGene results. Therefore, just QGene results using CIM are presented here. The detected QTLs individually accounted for 6.2–30.0% of the phenotypic variation. Of all the 35 QTLs, seven accounted for more than 10% of the phenotypic variation. Detected QTLs were named according to the nomenclature suggested by McCouch et al. (1997) and McCouch and CGSNL (2008). Here the letter ‘s’ for QTL nomenclature was used for a QTL mapped under saline conditions. A list of putative QTLs flanked by markers along with their LOD values, phenotypic variance and additive and dominance effects appears in table 3. The locations of all identified QTLs on 10 chromosomes of rice are illustrated in figure 2.
Number of days to flowering (DFL): QTL analysis using CIM identified three QTLs on chromosomes 4, 6 and 10 designated as qDTF4.1s, qDTF6.1s and qDTF10.1s, respectively (table 3). qDTF10.1s mapped to the interval RM271–RM484 on chromosome 10 with the largest effect explaining 13.3% of the total phenotypic variance. The Sadri allele at qDTF10.1s increased days to flowering by 4.8 days. The other two QTLs, qDTF4.1s and qDTF6.1s, explained 6.9 and 9.1% of the total phenotypic variance and the FL478 allele at these QTLs increased days to flowering by two and three days, respectively. qDTF4.1s exhibited dominance effects and increased DFL by FL478 allele. qDTF6.1s exhibited dominance effects and increased DFL by the Sadri allele.
Plant height (PHt): Four loci significantly associated with plant height were detected on chromosomes 1, 2, 3 and 7 using CIM and designated as qPH1.1s, qPH2.1s, qPH3.1s and qPH7.1s, respectively (table 3). The loci had R 2 values ranging from 6.6 to 17% and qPH1.1s, which was detected near marker RM212 located on the long arm of chromosome 1 had the highest value. The alleles from Sadri increased plant height at qPH1.1s, qPH3.1s and qPH7.1s, while alleles from FL478 increased plant height at qPH7.1s.
Panicle length (PnL): Three QTLs were detected on chromosomes 1, 2 and 3 for PnL. qPL3.1s, with the largest effect, explained 8.2% of the total phenotypic variation, and the additive effect of the Sadri allele increased PnL by 1.3 cm. qPL3.1s was mapped near marker RM130 located on the long arm of chromosome 3. The other two QTLs, qPL1.1s and qPL2.1s, explained 6.8 and 7.2% of the total phenotypic variance, respectively, and the Sadri alleles at these QTLs increased PnL (table 3).
Number of panicles per plant (PnN): Three QTLs for PnN were detected on chromosomes 4, 6 and 9. qPN6.1s had the highest R 2 value (9.2) was mapped to the interval RM20224–RM528 on chromosome 6 and the allele from Sadri increased the number of panicles. The other two QTLs, qPN4.1s and qPN9.1s, accounted for 8.6 and 7.9% of the phenotypic variation, with the FL478 alleles providing a positive effect on number of panicles (table 3). There is a big gap in qPN4.1s intervals, and we need to add more markers in this region to fill this gap.
Straw dry weight per plant (StDW): Four QTLs for straw dry weight were mapped on chromosomes 4, 7, 8 and 9, with R 2 values between 6.2 and 9.6. Among them, qSTW4.1s contributed 9.6% of the total phenotypic variation, with the FL478 allele providing an increased effect of 5.39 g. qSTW7.1s and qSTW9.1s, with the FL478 alleles, also had an increased effect on straw dry weight. qSTW8.1s contributed 6.2% of the total phenotypic variation, with the Sadri allele providing a positive effect of 5.05 g.
Number of fertile spikelets per plant (FrSp): In total, four QTLs were detected for number of fertile spikelets. They were designated as qFRSP2.1s, qFRSP4.1s, qFRSP6.1s and qFRSP10.1s. qFRSP2.1s was identified in the region of RM423–RM174 on chromosome 2, and explained 6.3% of the total phenotypic variation. Another QTL, qFRSP6.1s was mapped near marker RM275 on chromosome 6 and contributed 7.7% of the total phenotypic variation. The Sadri alleles had an additive effect at qFRSP2.1s and qFRSP6.1s loci of 110 and 108 fertile spikelets, respectively. The other two QTLs on chromosomes 4 and 10 accounted for 8.3 and 5.9% of the phenotypic variation. The FL478 alleles had a positive effect at these two loci by 105 and 114 spikelets, respectively. The dominant effects for qFRSP4.1s were increased FrSp by the Sadri allele (table 3).
Number of sterile spikelets per plant (StSp): Two QTLs were detected for number of sterile spikelets. qSTSP3.1s was detected near marker RM282 located on chromosome 3 and accounted for 9.2% of the phenotypic variation. The Sadri allele had an additive effect of 285 sterile spikelets. The other QTL, qSTSP7.1s, located on chromosome 7 near RM11, accounted for 7.1% of the total phenotypic variation. The FL478 allele had an additive effect of 47 sterile spikelets (table 3).
Number of total spikelets per plant (TSp): Three QTLs were detected for TSp on chromosomes 4, 7 and 9, collectively accounting for 27% of the total phenotypic variation. The larger-effect QTL, qTSP4.1s, flanked by RM551 and RM518 on chromosome 4, explained 12.4% of the variation. qTSP7.1s mapped in the region of RM445–RM11 on chromosome 7 contributed 7.8% of the phenotypic variation. Another QTL, qTSP9.1s, mapped near marker RM410 on chromosome 9, contributed 6.9% of the phenotypic variation. The alleles of these QTLs, which increased the number of total spikelets, came from FL478 (table 3).
Grain yield per plant (GYld): Four QTLs were detected for grain yield on chromosomes 2, 4, 6 and 8 and in total explained 31.6% of the phenotypic variation. The GYld QTLs under salinity stress were designated as qGY2.1s, qGY4.1s, qGY6.1s and qGY8.1s, respectively (table 3). qGY2.1s was detected in the region of RM555–RM324 located on chromosome 2 and accounted for 6.9% of the phenotypic variation. qGY6.1s mapped near marker RM275 on chromosome 6 contributed 8.4% of the phenotypic variation. The Sadri alleles at qGY2.1s and qGY6.1s increased grain yield by 3.2 g per plant. The other two QTLs affecting grain yield per plant, qGY4.1s and qGY8.1s, were identified on chromosomes 4 and 8, and accounted for 7.9 and 8.4% of the variation. The FL478 alleles by additive effects could increase grain yield at these two loci by 2.7 and 3.6 g per plant, respectively (table 3).
Spikelet fertility per cent (SpFr): Only a single QTL for spikelet fertility was detected near the marker RM555 located on chromosome 2 through CIM. This QTL, qSPFR2.1s, accounted for 7.9% of the phenotypic variation. The allele from Sadri could increase SpFr by 9.3%. While using IM, two more QTLs were detected on chromosomes 5 and 10 for spikelet fertility (table 3).
1000-grain weight (TGW): Two QTLs for TGW were detected, collectively explaining 61.4% of the phenotypic variation (table 3). A major QTL, qTGW8.1s, with a very large effect was detected under salt stress for TGW. This QTL was located within the RM80–RM281 region on chromosome 8 and accounted for 30% of the total phenotypic variation, and the FL478 allele had an additive effect of 2.95 g to increase TGW. The other large-effect QTL, qTGW10.1s, flanked by RM271 and RM484 on chromosome 10, accounted for 17.6% of the total variation. The FL478 allele could increase TGW by 2.36 g. The other two QTLs for 1000-grain weight, qTGW5.1s and qTGW6.1s, were identified on chromosomes 5 and 6 and accounted for 7.6 and 6.2% of the phenotypic variation. The Sadri alleles could increase TGW at both loci by 1 g.
QTL clusters: Four QTL clusters were observed on chromosomes 2, 4, 6 and 9. The first QTL cluster, flanked by RM423 and RM174 on the short arm of chromosome 3, contained three QTLs for FrSp, SpFr and GYld (figure 1). The allele from Sadri at these loci increased FrSp, SpFr and GYld. The second QTL cluster was flanked by RM551 and RM518 on the short arm of chromosome 4, including qSTW4.1s, qFRSP4.1s, qFRSP4.1s, qTSP4.1s and qGY4.1s. The FL478 allele of these QTLs could increase StDW, FrSp, TSp and GYld. The third QTL cluster was located in the interval of RM20224–RM528 on the long arm of chromosome 6. It contained QTLs for PnN, FrSp, GYld and TGW (figure 1). The Sadri allele had a positive effect on all four traits. The fourth QTL cluster was associated with RM242 on chromosome 9, including qPN9.1s, qSTW9.1s and qTSP9.1s. The allele from FL478 at this region could increase PnN, StDW and TSp. This region has been reported as a QTL cluster for yield components (Xie et al. 2008).
QTLs for correlated traits were often mapped in the same chromosomal regions (Veldboom et al. 1994). This trend was observed in our study. For example, StDW and GYld were correlated and had two QTLs, qSTW4.1s and qGY4.1s, which were found at the same map locations on chromosome 4. TGW and GYld also showed a high correlation and had two QTLs, qTGW8.1s and qGY8.1s, in a similar location on chromosome 8 (table 2). In both cases, the directions of the correlations were consistent with that of the effects of the QTLs on the traits. These colocalized QTLs could be attributed to pleiotropy or tightly linked genes.
The significant threshold of each putative QTL detected by CIM was reconfirmed using 10,000 permutations for the presence of each QTL across the 12 chromosomes (Churchill and Doerge 1994). The significance level of the detected QTLs had average LOD scores greater than 3.6 with P < 0.05. Using permutations, 28 QTLs out of the 35 detected QTLs were reconfirmed. QTLs reconfirmed through these permutations may potentially be useful as fine-mapping targets for map-based gene cloning and functional analysis and also for breeding by marker-assisted backcrossing (MABC).
Discussion
Both parental lines, Sadri and FL478, contributed additive effects for QTLs for days to flowering, plant height, number of panicles, straw dry weight, number of fertile spikelets, number of sterile spikelets, grain yield per plant and 1000-grain weight. Sadri contributed additive effects in the QTLs for panicle length and spikelet fertility, while FL478 contributed additive effects in the QTLs for total spikelet number. A majority of the QTLs detected in our study are reported for the first time for the yield components of rice under reproductive stage salinity stress.
Moreover, identifying additional salt tolerance-related QTLs from different donors would give rice breeders a wider option in combining superior QTLs into one genetic background using gene-pyramiding techniques, since the commonly used donor parents for salinity tolerance seem to possess a few superior tolerance traits (Ismail et al. 2007). Combining genes for tolerance at all developmental stages will facilitate the development and release of new robust rice varieties with substantially higher salt tolerance (Thomson et al. 2010). Most QTL mapping studies have been limited to seedling-stage tolerance because phenotyping for salinity tolerance at reproductive stage is very tedious and time-consuming. There have been only few reports on QTL analysis of rice salt tolerance at the reproductive stage but mostly unpublished (Singh and Flowers 2010). Islam (2004) evaluated 80 RILs of IR29/Pokkali at the reproductive stage using salinized water (EC = 5 dS m − 1) at the IRRI greenhouse and reported a QTL on short arm of chromosome 4 for per cent reduction for biomass weight and another QTL for per cent reduction in filled grain weight on chromosome 7 flanked by RM445 and RM11. In our study, a QTL was mapped on the short arm of chromosome 4 for straw dry weight, which is similar to the finding of Islam (2004). Palao et al. (Palao C. D. C., Vina C. B., Gregorio G. B., Thomson M. J. and Singh R. K. 2012 Identification of major QTLs for salinity tolerance at the reproductive stage in rice (Oryza sativa L.), Abstract in the first IRRI Young Scientists Conference, IRRI, Los Baños, Philippines, 8–9 November 2012) used 201 F2 plants from IR64/IR4630-22-2-5-1-3 for QTL mapping under salinity stress at the reproductive stage in rice in greenhouse conditions. F2 plants were exposed to salinity stress at ECe 10 dS m − 1 before the booting stage. Thirty-four QTLs were detected for traits such as plant height, panicle length, single-grain weight, straw weight, root dry weight, %Na, %K and Na–K ratio. But their study found some unique QTLs for plant height, panicle length and straw weight, which did not coincide with the QTLs found in our study. This may be attributed to the different populations used and screening conditions employed.
Our study supported claims from previous studies that the genes governing salinity tolerance at the seedling and reproductive-stage are very different. No QTLs were detected on the short arm of chromosome 1 where the Saltol QTL and SKC1 have been reported. This reconfirms that the genes and QTLs underlying the mecahnism for seedling and reproductive stages salinity are very different and pooling of both mechanisms in one background could be a possible answer for salinity tolerance throughout crop growth with sustainable grain yield.
The Sadri/FL478 F2 population showed a good combination of seedling and reproductive stage salinity tolerance. This population has seedling and reproductive-stage tolerance genes from FL478 and reproductive-stage tolerance genes from Sadri. The F3 population of Sadri/FL478 is currently available. This population will be screened under salinity stress at both the seedling and reproductive stages and salt tolerant progenies will proceed to advanced generations to breed salt-tolerant rice lines. The advanced population could be used to validate QTL effects for MABC. Fine-mapping of selected QTLs will help to identify closely linked markers for use in MABC to supplement the breeding programmes.
The knowledge of QTLs for salt tolerance will be a valuable tool in future plant breeding programmes for producing high-yield rice varieties for salt-affected environments. Pyramiding salt-tolerance QTLs using MAS will be useful for the development of new varieties with high salt tolerance for salinity-affected regions (Lin et al. 2004). QTLs identified for both the seedling and reproductive stages could be used to combine genes controlling different physiological mechanisms into a single genetic background to enhance salt tolerance of rice.
References
Akbar M., Yabuno T. and Nakao S. 1972 Breeding for saline resistant varieties of rice I. variability for salt tolerance among some rice varieties. Jpn. J. Breed. 22, 277–284.
Ammar M. H. M., Pandit A., Singh R. K., Sameena S., Chauhan M. S., Singh A. K. et al. 2009 Mapping of QTLs controlling Na + , K + and Cl − ion concentrations in salt tolerant indica rice variety CSR27. J. Plant Biochem. Biotech. 18, 139–150.
Churchill G. A. and Doerge R. W. 1994 Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971.
Counce P. A., Keisling T. C. and Mitchell A. J. 2000 A uniform, objective, and adaptive system for expressing rice development. Crop Sci. 40, 436–443.
Cui H., Takeoka Y. and Wada T. 1995 Effect of sodium chloride on the panicle and spikelet morphogenesis in rice. Jpn. J. Crop. Sci. 64, 593–600.
Flowers T. J. and Yeo A. 1981 Variability in the resistance of sodium chloride salinity within rice (Oryza sativa L.) varieties. New Phytol. 88, 363–373.
Flowers T. J. and Yeo A. 1995 Breeding for salinity resistance in crop plants—where next? Aust. J. Plant Physiol. 22, 875– 884.
Flowers T. J. and Flowers S. A. 2005 Why does salinity pose such a difficult problem for plant breeders? Agric. Water Manage. 78, 15–24.
Genc Y., McDonald G. and Tester M. 2007 Reassessment of tissue Na + concentration as a criterion for salinity tolerance in bread wheat. Plant Cell Environ. 30, 1486–1498.
Gregorio G. B. 1997 Tagging salinity tolerance genes in rice using amplified fragment length polymorphism (AFLP). Ph.D. thesis, University of the Philippines, Los Ba nos College, Laguna, Philippines.
Gregorio G. B., Senadhira D., Mendoza R. D., Manigbas N. L., Roxas J. P. and Guerta C. Q. 2002 Progress in breeding for salinity tolerance and associated abiotic stresses in rice. Field Crops Res. 76, 91–101.
Haq T. U., Gorham J., Akhtar J., Akhtar N. and Steele K. A. 2010 Dynamic quantitative trait loci for salt stress components on chromosome 1 of rice. Funct. Plant Biol. 37, 634– 645.
Heenan D. P., Lewin L. G. and McCaffery D. W. 1988 Salinity tolerance in rice varieties at different growth stages. Aust. J. Exp. Agric. 28, 343–349.
Islam M. M. 2004 Mapping salinity tolerance genes in rice (Oryza sativa L.) at reproductive stage. Ph.D. dissertation, University of the Philippines, Los Ba nos, Laguna, Philippines.
Ismail A. M., Heuer S., Thomson M. J. and Wissuwa M. 2007 Genetic and genomic approaches to develop rice germplasm for problem soils. Plant Mol. Biol. 65, 547–570.
Jenks M. A., Hasegawa P. M. and Jain S. M. 2007 Advances in molecular breeding toward drought and salt tolerant crops. Springer, The Netherlands.
Joehanes R. and Nelson J. 2008 QGene 4.0, an extensible Java QTLanalysis platform. Bioinformatics 24, 2788–2789.
Khatun S., Rizzo C. A. and Flowers T. J. 1995 Genotypic variation in the effect of salinity on fertility in rice. Plant Soil 173, 239–250.
Kim S.-H., Bhat P. R., Cui X., Walia H., Xu J., Wanamaker S. et al. 2009 Detection and validation of single feature polymorphisms using RNA expression data from a rice genome array. BMC Plant Biol. 9, 65.
Kosambi D. D. 1944 The estimation of map distances from recombination values. Ann. Eugen. 12, 172–175.
Lin H. X., Zhu M. Z., Yano M., Gao J. P., Liang Z. W., Su W. A. et al. 2004 QTLs for Na + and K + uptake of the shoots and roots controlling rice salt tolerance. Theor. Appl. Genet. 108, 253–260.
Maas E. V. and Hoffmann G. J. 1977 Crop salt tolerance: current assessment. J. Irrig. Drain. Eng.-ASCE. 103, 115–134.
Maas E. V. and Grattan S. R. 1999 Crop yields as affected by salinity. In Agricultural drainage (ed. R. W. Skaggs and J. van Schilfgaarde), pp. 55–108. Agron Monograph 38, ASA, CSSA, SSA, Madison, USA.
Manly K. F., Cudmore Jr R. H. and Meer J. M. 2001 Map Manager QTX, cross-platform software for genetic mapping. Mamm. Genome 12, 930–932.
McCouch S. R. and CGSNL (Committee on Gene Symbolization, Nomenclature and Linkage, Rice Genetics Cooperative) 2008 Gene nomenclature system for rice. Rice 1, 72–84.
McCouch S. R., Chen X., Panaud O., Temnykh S., Xu Y., Cho Y. G. et al. 1997 Microsatellite marker development, mapping and applications in rice genetics and breeding. Plant Mol. Biol. 35, 89–99.
Mohammadi-Nejad G., Arzani A., Rezai A. M., Singh R. K. and Gregorio G. B. 2008 Assessment of rice genotypes for salt tolerance using microsatellite markers associated with the saltol QTL. Afr. J. Biotechnol. 7, 730–736.
Moradi F., Ismail A. M., Gregorio G. B. and Egdane J. A. 2003 Salinity tolerance of rice during reproductive development and association with tolerance at the seedling stage. Ind. J. Plant Physiol. 8, 105–116.
Munns R. and Tester M. 2008 Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681.
Narciso J. and Hossain M. 2002 World rice statistics. IRRI, Los Ba nos, Philippines.
Rao P. S., Mishra B., Gupta S. R. and Rathore A. 2008 Reproductive stage tolerance to salinity and alkalinity stresses in rice genotypes. Plant Breed. 127, 256–261.
Ren Z. H., Gao J. P., Li G. L., Cai X. L., Huang W., Chao D. Y. et al. 2005 A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet. 37, 1141–1146.
Sarhadi E., Mohammadi-Bazargani M., Sajise A. G., Abdolahi S., Vispo A. N., Arceta M. et al. 2012 Proteomic analysis of rice anthers under salt stress. Plant Physiol. Biochem. 58, 280–287.
Singh R. K. and Flowers T. J. 2010 The physiology and molecular biology of the effects of salinity on rice. In Handbook of plant and crop stress, 3rd edition (ed. M. Pessarakli), pp. 901–942. Taylor and Francis, Boca Raton, USA.
Singh R. K., Gregorio G. B. and Jain R. K. 2007 QTL mapping for salinity tolerance in rice. Physiol. Mol. Biol. Plants 13, 87–99.
Temnykh S., DeClerck G., Lukashova A., Lipovich L., Cartinhour S. and McCouch S. R. 2001 Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 11, 1441–1452.
Thomson M. J., Edwards J. D., Septiningsih E. M., Harrington S. E. and McCouch S. R. 2006 Substitution mapping of dth1.1, a flowering-time quantitative trait locus (QTL) associated with transgressive variation in rice, reveals multiple sub-QTL. Genetics 172, 2501–2514.
Thomson M. J., Ocampo M., Egdane J., Rahman M. A., Sajise A. G., Adorada D. L. et al. 2010 Characterizing the Saltol quantitative trait locus for salinity tolerance in rice. Rice 3, 148–160.
United States Salinity Laboratory Staff 1954 Diagnosis and improvement of saline and alkali soils. US Department of Agriculture, Agricultural Handbook No. 60. Government Printer, Washington, USA.
Veldboom L. R., Lee M. and Woodman W. L. 1994 Molecular-marker facilitated studies in an elite maize population. I. Linkage analysis and determination of QTLs for morphological traits. Theor. Appl. Genet. 88, 7–16.
Wang S., Basten C. J. and Zeng Z. B. 2011 Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, USA. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Xie X., Jin F., Song M. H., Suh J. P., Hwang H. G., Kim Y.G. et al. 2008 Fine mapping of a yield-enhancing QTL cluster associated with transgressive variation in an Oryza sativa L. × O. rufipogon cross. Theor. Appl. Genet. 116, 613–622.
Yeo A. R. and Flowers T. J. 1982 Accumulation and localization of sodium ions within the shoots of rice (Oryza sativa) varieties differing in salinity resistance. Physiol. Plant 56, 343–348.
Zang J. P., Sun Y., Wang Y., Yang J., Li F., Zhou Y. L. et al. 2008 Dissection of genetic overlap of salt tolerance QTLs at the seedling and tillering stages using backcross introgression lines in rice. Sci. Chin. Ser. C-Life Sci. 51, 583–591.
Zeng L. and Shannon M. C. 2000 Salinity effects on the seedling growth and yield components of rice. Crop Sci. 40, 996–1003.
Zeng L., Shannon M. C. and Grieve C. M. 2002 Evaluation of salt tolerance in rice genotypes by multiple agronomic parameters. Euphytica 127, 235–245.
Zeng Z. 1993 Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad Sci. USA 90, 10972–10976.
Zeng Z. 1994 Precision mapping of quantitative trait loci. Genetics 136, 1457–1468.
Zheng L., Shannon M. C. and Lesch S. M. 2001 Timing of salinity stress affecting rice growth and yield components. Agric. Water Manage. 48, 191–206.
Acknowledgements
The authors thank A. G. Sajise and N. A. Vispo for technical assistance in the salinity screening, and M. Arceta for technical assistance in the genotyping. We also appreciate Dr Endang M. Septiningsih and Dr Bert C. Y. Collard for their useful suggestions and comments. Technical editing by Dr Bill Hardy is sincerely acknowledged. The help of the head of the Plant Breeding, Genetics and Biotechnology Division is duly acknowledged for permission to conduct the research at IRRI.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Mohammadi R., Mendioro M. S., Diaz G. Q., Gregorio G. B. and Singh R. K. 2013 Mapping quantitative trait loci associated with yield and yield components under reproductive stage salinity stress in rice (Oryza sativa L.). J. Genet. 92, xx–xx]
Rights and permissions
About this article
Cite this article
MOHAMMADI, R., MENDIORO, M.S., DIAZ, G.Q. et al. Mapping quantitative trait loci associated with yield and yield components under reproductive stage salinity stress in rice (Oryza sativa L.) . J Genet 92, 433–443 (2013). https://doi.org/10.1007/s12041-013-0285-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-013-0285-4