
Abstract
1,2,3-Triazole and tetrazole derivatives bearing pyrrolidines are found to exhibit notable biological activity and have become useful scaffolds in medicinal chemistry for application in lead discovery and optimization. We report design, synthesis and molecular docking studies of tetrazolyl-1,2,3-triazole derivatives (7a-i) bearing pyrrolidine moiety and evaluating their anticancer activity against four cancer cell lines viz. Hela, MCF-7, HCT-116 and HepG2. The structures of the new compounds were ascertained by spectral means IR, NMR: 1H &13C and Mass spectrum. From the studies compounds7a and 7i exhibited significant anticancer activity against the Hela cell line with IC50 = 0.32 ± 1.00, 1.80 ± 0.22 μM when compared to reference drug Doxorubicin (IC50 = 2.34 ± 0.11 μM), whereas 7h, 7i, and 7b were found to be active against MCF-7, HCT-116 and HepG2 cell lines with IC50 = 3.20 ± 1.40, 1.38 ± 0.06 and 0.97 ± 0.12 μM respectively. Notably 7a exhibited highest conventional hydrogen bondings TyrA:40, SerA:17, LysA:117, AlaA:146, Tyr218 with 3HB4and SerA:17, LysA:117, AlaA:146, TyrA:40 with 6IBZ and docking energy − 10.85, − 8.21 kcal/mol respectively. These compounds were further evaluated for their ADMET and physicochemical properties by using SwissADME. The results of the in vitro and in silico studies suggest that the tetrazole incorporated pyrrolidine-triazoles may possess the ideal structural requirements for further developing new anticancer agents.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The uncontrolled growths of abnormal cells lead to cause deadly diseases. Cancer is the second biggest cause of illness and mortality after heart disorders according to the World Health Organization. Though there is tremendous growth in the screening of various types of cancer, emergence of effective drugs and prevention, cancer is still the leading cause of death in people [1]. Most of the currently used chemotherapeutic drugs are ineffective because of the development of drug resistance during treatment, in spite of advances in the understanding of the molecular biology of cancer and the ensuing rise in the development of anticancer compounds [2]. According to GLOBOCAN 2018, approximately 18.1 million new cases of cancer have been recognized and among them, lung cancer (18.4%), followed by breast (11.6%), prostate (7.1%), colorectal (6.1%), stomach and liver cancer are the most common [3]. Breast cancer is the most frequently diagnosed cancer and the leading cause of death among females, accounting for 23% of the total cancer cases and 14% of cancer deaths; thus, research in this field is important to overcome both economical and psychological burden [4]. As for breast cancer, MCF-7 cells represent a very important candidate as they are used ubiquitously in research for estrogen receptor (ER)-positive breast cancer cell experiments and many sub-clones, which have been established, represent different classes of ER-positive tumours with varying nuclear receptor expression levels [5]. The second most frequent female malignant tumour in the world, cervical cancer poses a major threat to the health of women [6]. HeLa cell line was a particularly aggressive strain of cervical cancer cells acquired during a normal biopsy from a 30-year-old mother of five. It has been determined that persistent infection of high-risk human papillomavirus (HPV) is the cause of cervical cancer. Therefore, the need for new and effective anticancer agents increased for the treatment of new increasing number of cancer patients [3].
5-membered heterocycles with nitrogen are structural motifs that have undergone substantial research in the creation of molecules with a range of biological potential. The tetrazole pharmacophore is present in several current medications [7, 8], in particular, 1,5-substituted tetrazole derivatives have been employed as peptidomimetics and have a wide range of applications in medicinal chemistry as substitutes for cis-amide linkages [9]. Additionally, tetrazole-bearing derivatives are claimed to have biological properties that are anti-hypertensive [10], antibacterial [11], anticonvulsant [12], analgesic[13], antiproliferative [14], anti-fungal [15], anti-tuberculosis[16], anti-malarial, anti-leishmaniosis [17], anti-diabetic [18], anticancer [19], and many other biological activities. Another well-known heterocycles 1,2,3-triazoles, which have a wide range of biological applications, including anticonvulsant [20], antiviral [21], anti-tubercular [22], antibacterial [23], antifungal [24], anticancer [25], antioxidant [26], antimalarial [27], anti-alzheimer effect [28], antidepressant [29], anti-inflammatory [30], antiplatelet [31], anti-HIV activities [32]. Because of their outstanding biological activity profiles, these heterocyclic compounds are well documented in the literature for the treatment of various diseases and deadly tumours as shown in (Fig. 1). Based on the information above and ongoing work, we have designed a small number of hybrid molecules that combine the pyrrolidine, tetrazole, and 1,2,3-triazole pharmacophores in a single framework.

Chemical structures of the reported anticancer active molecules bearing pyrrolidine, 1,2,3-triazole and tetrazole
Additionally, the pharmacokinetic properties of the final 1,2,3-triazolyl tetrazoles (7a–i) were predicted using absorption, distribution, metabolism excretion, and toxicity (ADMET) descriptors by a SwissADME (https://www.swissadme.ch/), ADMETlab2.0 (https://admetmesh.scbdd.com server. In silico ADMET is currently used widely to determine whether it is possible for a drug candidate to reach its site of action. The synthesized compounds may have fascinating biological features that are effective against many cancer cell lines. To our knowledge, the targeted pyrrolidine-tetrazoles 7a–i and the applied methodology (Fig. 2) are not reported previously. Therefore, we performed the synthesis and docking studies of tetrazole–triazole linked pyrrolidine derivatives, and these results suggested that they might be lead compounds for treating cancer infections that are resistant to treatment.

Rational design strategy of tetrazole incorporated pyrrolidine–triazoles, a structure of the previously reported Anticancer compound with pyrrolidine ring, b example of 1,2,3-triazole derivative used in Anticancer therapy, c representative example of tetrazole molecule with sulfur that exhibits Anticancer activity
Results and discussion
Chemistry
In the present study, we reported the synthesis and structural characterization of a novel series of tetrazolyl 1,2,3-triazoles bearing pyrrolidines and their derivatives as lead compounds via 4-steps. (Scheme 1, Fig. 3). Compounds 2,4-difluoro-3-methoxybenzoyl chloride 1 and (2S,4R)-methyl-4-hydroxypyrrolidine-2-carboxylate hydrochloride 2 were synthesized by following the previously reported synthetic procedures and detailed experimental procedures are provided in the supporting information [33, 34]. Synthesis of key intermediate (2S,4R)-methyl 1-(2,4-difluoro-3-methoxybenzoyl)-4-hydroxy pyrrolidine-2-carboxylate 3 was attained by following the reaction conditions reported in the literature with slight modifications [34]. (2S,4R)-methyl-4-hydroxypyrrolidine-2-carboxylate hydrochloride 2 was suspended in pyridine and triethylamine for 20 min at ambient temperature.

Synthetic pathway of tetrazole–triazole linked pyrrolidine derivatives

List of synthesized tetrazole–triazole linked pyrrolidine derivatives with yield
Then it was reacted with 2,4-difluoro-3-methoxy benzoyl chloride 1 in dichloromethane for 4 h, followed by filtration and dried to provide pale yellow solid (key intermediate 3, 75%). Intermediate 3 was reacted with 4-tolylsulfonyl chloride in dichloromethane and triethylamine to provide Tosylate 4 as a white crystalline solid. The Tosylate was treated with sodium azide in dry DMF at 50–55 °C to provide pale yellow oil, which solidified upon standing to provide the azide intermediate 5 (SN2 reaction with sodium azide and resulted in the inversion of stereocenter, 83%). A series of 1-phenyl-5-(prop-2-yn-1-ylthio)-1H-tetrazoles 6a–i were synthesized as per the reported literature starting from phenyl isothiocyanate which was reacted with sodium azide to give 1-phenyl-1H-tetrazole-5-thiol [35]. Then the solution of 1-phenyl-1H-tetrazole-5-thiol, propargyl bromide, and tetrabutylammonium bromide in a mixture of triethylamine and dichloromethane was stirred for 4 h to obtain the tetrazole derivatives (80–87%). The synthesis of new tetrazolyl triazole pyrrolidine derivatives was accomplished via Click chemistry of appropriate substituted tetrazole alkynes 6a–i with pyrrolidine azide 5, L-sodium ascorbate in mixture of DMF-H2O stirred for 4–5 h at room temperature. The synthesized tetrazolyl triazole derivatives 7a–i were obtained in good yields of up to 92%.
Spectral analysis
All the synthesized compounds 7a–i were characterized by spectral techniques prior to screening for anticancer activity. The IR spectrum of 7a has shown the characteristic absorption bands for = CH (aromatic), − CH (alkanes) at 3037.75, 2927 cm−1 respectively. Two strong peaks at 1699.86 cm−1 and 1637.56 cm−1 were pertaining to stretching frequency of the carbonyl group (C=O); the higher frequency band was allocated to ester carbonyl and the lower frequency band was allocated to amide carbonyl vibrations. The bands at 1597.22, 1514.65 cm−1 and1406.41 cm−1 being assigned to the valence bonds Ar−C=C and − C=N groups. The C–S–C, C–O–C and C–F functional groups were characterized by the presence of the vibrational peaks at 1209.47 cm−1, 1103.49 cm−1 and 1040.23 cm−1 respectively. The 1H NMR spectrum of compound 7a exhibited two doublets of doublets at δ 7.37, 8.03 ppm and three doublets at 6.81, 7.54, and 8.44 ppm for aromatic protons respectively. The singlet at 8.62 ppm could be attributed to the triazole proton, whereas the peaks at 3.77, 2.88 and 4.88 ppm were assigned to methoxy (Ar–OCH3& –C–OCH3) and –SCH2 groups respectively. The pyrrolidine moiety presents four types of protons, three multiplets, two doublets and a triplet. The triplet at δ 2.65 ppm is attributable to the proton CH whereas, the doublets at δ 3.31, 2.98 ppm are attributed to the protons CH2. The protons of the methylene moiety exhibited multiplet between δ 0.93–1.65 ppm overlapping with the protons of the adjacent nucleus. The 13C NMR of this compound has shown two carbonyl signals at δ 176.2, 172.0 ppm, which are assigned to the carbons of amide and ester groups. Moreover, the aromatic carbons revealed that the signals at δ 160.0, 161.4, 156.2 ppm were attributed to carbon phenolic fragment bearing fluoro benzene groups. Furthermore, one can notice an overlapping of the signals due to aromatic carbons in the range δ 102.5–142.3 ppm whereas the signals exhibited at δ 151.5, 150.0 ppm due to tetrazole, triazole carbons respectively. For compound 7a, the molecular ion peak m/z at 593 corresponding to [M + H]+ was observed by the mass spectrum (ESI–MS) form which the molecular weight is consistent with the molecular formula C24H20F4N8O4S. The chemical structures of all the synthesized compounds are presented in Fig. 3.
Anticancer activity
Anticancer activity of these compounds has been assessed in vitro by MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay against four Hela, MCF-7, HCT-116, and HepG2 cell lines [36,37,38]. The percentage of cell death was measured for the new pyrrolidines linked to dual heterocycles7exhibited higher anticancer activities at various concentrations along with IC50 values (Table 1, Fig. 4).Among the tested compounds7a, 7c, 7 h, and 7i showed excellent anticancer activity against both Hela (human cervix epithelioid carcinoma), MCF-7 (human breast adenocarcinoma) cell lines. From screening results compounds displayed promising activity and 7a, 7i exhibited the highest cytotoxicity against Hela cell line with IC500.32 ± 1.00, 1.80 ± 0.22 μM and 7f, 7c shows good activity against Hela cell line with IC504.83 ± 1.02, 4.85 ± 1.15 μM respectively. Compounds7a, 7h, 7i with difluorophenyl pyrrolidine tetrazole, orthohydroxyphenyl triazolyl-tetrazole, dihydroxyphenyl pyrrolidine elicited good activity against MCF-7 cell line with IC50 value 3.83 ± 0.37, 3.20 ± 1.40, 4.15 ± 0.55 μM respectively. Morover the compounds7c, 7dand 7f displayed moderate to good anticancer activity againstMCF-7 cell line with IC50 range 10.77 ± 1.12, 5.06 ± 0.17, 8.11 ± 2.12 μM respectively. Compounds 7i, 7b, 7a and 7e displayed excellent anticancer activity against human colon carcinoma (HCT-116) cell line with IC50 values of 1.38 ± 0.06, 1.48 ± 0.08, 1.66 ± 0.64, and 4.72 ± 0.03 μM where as the dual heterocyclic compounds 7fand 7gexhibitedthe superior anticancer activity against HCT-116 cell line with IC50 value of 10.73 ± 0.52, 11.90 ± 0.19 μM respectively. Preliminary cytotoxicity was performed with the synthetized heterocyclic compounds and the positive control, doxorubicin towards (human caucasian hepatocyte carcinoma) HepG2 cell line. All selected compounds 7b, 7a, 7i, and7e had cytotoxic effect in the tested hepatocyte carcinoma cell line with IC50 values 0.97 ± 0.12, 1.92 ± 0.06, 5.08 ± 1.49, and 3.25 ± 0.07 µM separately. In the present study, it was noted that dual heterocyclic compounds 7a, 7f, 7i, 7e, 7b, and 7 h had IC50 values below or around 10 µM on all the four selected cancer cell lines including drug-sensitive and multidrug-resistant phenotypes. Amongst them, all the title compounds and doxorubicin displayed IC50 values below 20 µM, 7b had an IC50 value of 23.17 ± 1.07 µM meanwhile other compounds 7b (Hela), 7c, 7h (HCT-116), 7f (HepG2), 7 g (MCF-7) were not active at up to 50 µM (Table 1).

A IC50 values for the most active pyrrolidines against human cervix epithelioid carcinoma (Hela),human breast adenocarcinoma (MCF-7), human colon carcinoma (HCT-116), and human caucasian hepatocyte carcinoma (HepG2) cell lines. B Rates of Inhibition of cell invasion by various concentrations of 7i(HCT-116).Data are mean ± SD (n = 5)
Regarding the structure–activity relationship, it appears that the synthesized 2,4-fluoro phenyl tetrazole derivative 7a and 2-chloro-4-fluoro phenyl tetrazolyl triazole derivative 7b had excellent or having good cytotoxic effects against Hela (IC50 values below 10 µM) (Fig. 5). In contrast, o-nitro 7f, 2, 5- dimethoxy pyrrolidines 7c with tetrazole and 1,2,3-triazole substituents has the best cytotoxic effect with IC50 values below 10 µM against Hela cancer cell line (Table 1). This is an indication that electron-donating substituents decrease the anticancer activity of pyrrolidines whereas electron-withdrawing substituents increase the activity against all the tested cell lines. Within the tetrazolyl triazole pyrrolidines, it was observed that the numbers and positions of both methoxy (7b) methyl (7d), and hydroxy (7 h) functional groups did not significantly influence the cytotoxic activity. The presence of fluoro (7a), chloro (7b) and nitro (7e) substituents significantly increases the anticancer activity against all the tested cell lines. However, the presence of nitro and hydroxy substituents at ortho position of the phenyl ring (7f & 7h) has minimized the biological activity, probably due to the steric hindrance that might prevent the compounds to reach its biological target.

SAR summary for anticancer activities of tetrazolyl-triazole pyrrolidine derivatives (7)
Molecular docking studies
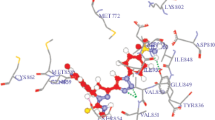
Breast and carcinoma cancer cell lines contain a diverse set of enzymes that are considered necessary for MCF-7, HCT-116 cell lines against17β-HSD type 1, PFKFB3 inhibitors and reproduction. One of these enzymes that have been recognized as an anticancer target is breast cancer and carcinoma cancer therapy. To validate the accuracy of Autodock 4.2 is an appropriate docking tool for the present purpose, the co-crystal structure of 17β-hydroxy steroid dehydrogenase type1 complexed with E2B (3HB4),and the co-crystallized structure of Human PFKFB3 in complex with a N-Aryl 6-Aminoquinoxaline inhibitor 7 (6IBZ)having 2.21 Å resolution[39, 40]. According to the method of validation cited in the literature, the successful scoring function is the one in which the RMSD of the best-docked conformation is ≤ 2.0 Å from the experimental one [41,42,43]. Pyrrolidines 7e, 7i, 7a, 7h have shown highest docking energies of − 11.83, − 11.50, − 10.85, − 9.28 kcal/mol with 17β-hydroxysteroid dehydrogenase protein and formed various π-interactions such as π−π, π-cation, sigma−sigma n − π and π-sigma, alkyl contacts (Table 2, Fig. 6). The ligand 7a displayed five conventional H-bondings TyrA:40 (O–F) (3.101 Å), SerA:17 (triazole pocket) (2.942 Å), LysA:117 (sulfur pocket) (2.016 Å), AlaA:146 (fluoro benzene pocket) (2.633 Å), Tyr218 (2.165 Å), three π-alkyl residues with ValA:29, AlaA:18 (b/n triazole and tetrazole), PheA:28, one π-anion residue with AspA:38, two fluorine interactions with AspA:119, AsnA:116. Whereas the new 1,2,3-triazolyl tetrazole 7a disclosed the diverse noteworthy hydrogen bonding interaction with the active site of amino acid radius SerA:17 (2.346 Å), LysA:117 (1.948 Å), AlaA:146 (2.795 Å), TyrA:40 (2.648 Å), and Π-cation, anion bondings with ArgA:75, AsnA:163 (F), CysA:154, GluA:166. For a most active compound 7e reveals a dissociation constant of 84.30 nM and exhibited the highest hydrogen binding modes with LysA:147, AlaA:146, LysA:117, SerA:17, AsnA:116at 1.348, 1.678, 2.037,1.975and 2.084 Å respectively. Moreover, this compound possesses carbon hydrogen bondings with AspA:33, ProA:34, ThrA:35, one π-π interaction with PheA:28 (2.348 Å), and two Pi-alkyl residues ValA:29, TyrA:40 against 6IBZ protein. Whereas the ligand 7e reveals significant H-bond interactions withGlyX:15 (2.112 Å), ValX:143 (2.120 Å), Ile:14 (2.004 Å), and notableπ-cation, π-alkyl interactions like LysX: 159, IleX:14, ValX:225, PheS:259 the hydrogen bonding stackings (Green), carbon-hydrogen bond (Aqua), π–π (pink), Pi-halogen (blue), π-anion (dark orange), π-sigma (violet), π-sulphur (yellow), π-alkyl and sigma stackings(rose) were found. The most important derived ligand 7i exhibits seven stronger H-bond interactions with amino acidLysX:159, AsnX:90, TyrX:155, GlyX:92, SerX:142, GlyX:144, GlyX:94 with bond distance 2.183, 2.165, 2.013, 1.326, 2.015, 1.954, 2.103 Å and the other interactions like carbon-hydrogen bondings exhibitsGlyX:186, CysX:185, ProX:187, one Pi-Pi bonding PheX:192, and four π-alkyl bondings with amino acids MetX:193, ValX:196, ValX:143, LeuX:149 against 3HB4. Compound 7i formed π–π, n–π, π-alkyl amino acids AspA:33, AlaA:146, LysA:147, PheA:28, LysA:117, AlaA:18, TyrA:32 with substituted aromatic ring and pyrrolidine pocket residue respectively and also formed a hydrogen binding interactions with SerA:17 (2.018 Å), ProA:34 (2.394 Å), AspA:38 (2.236 Å), TyrA:40 (2.102 Å) via sulfur, phenol, tetrazole & phenol, tetrazole substituents in the active site of Human PFKFB3 in complex with an N-aryl 6-aminoquinoxaline inhibitor (Fig. 7). Based on the docking energies, the tested compounds have more effective inhibitors in following PFKFB3, E2B receptor interactions with modern drug design.

Validation of the docking study into active site of proteins 3HB4; A) 2D-structures of 7b, 7e, 7i, DXN; B)3D-side chine flexibility of 7b, 7e, 7i, DXN against human breast adenocarcinoma (3HB4). Probable binding mode of compounds (lime color) in the active site of a subunit from M. tuberculosis; the hydrogen bond interactions are shown in light green color dotted lines, the important residues are shown in cyan/aqua color, and protein cartoon is represented according to the secondary structure of the protein

A 2D-structures of 7b, 7e, and 7i against human colon carcinoma (6IBZ)
ADMET predictions
Drug-likeness assessment
Further investigation, these analogues have been predicted for their drug-likeness scores and pharmacokinetic properties including ADMET parameters to verify that the designed molecules are viable drugs (Table 3, Fig. 8). Ghose, Veber, Egan, and Muegge filters, as well as Lipinski's rule of five, were not projected to be satisfied by the chosen compounds [44]. They had good lipophilic properties, a consensus log Po/w value of 3.89, a high GI absorption score (17%), and were expected not to pass through the blood–brain barrier (BBB) or act as a P-gp substrate. The designed compounds are inhibitors of the cytochrome P450 isoenzymes CYP1A2 and CYP2C19, but not of CYP2D6 or, more specifically, CYP3A4, which is known to be the most abundant in the liver and an active participant in the metabolism of about 45% of known drugs [45, 46]. This was revealed by predictive data. The chosen compounds' skin permeation rates (Log KP) ranged from − 8.44 to − 7.54 cm/s. Brain Or Intestinal estimated permeability approach (BOILED-Egg) was presented as an effective prediction model based on small molecule lipophilicity and polarity calculations in order to attain this objective. The BOILED-Egg model offers a rapid, simple, easily reproducible, and statistically unmatched method for predicting the excellent gastrointestinal absorption and brain permeability of tiny compounds that may be used in drug development and discovery. The white part of the egg (the yolk) seems to be the physicochemical zone of substances that are likely to be absorbed by the GI tract. The Blood–Brain Barrier, shown in yellow, is a physical and chemical zone where chemicals that are likely to get into the brain (BBB permeation) are kept. As shown in Fig. 8, the compound7b with apparent oral bioavailability was superimposed over the BROILED-Egg. According to medicinal chemistry qualities, none of the compounds displayed any pain or brink alerts, and the bulk of them had generally good synthetic accessibility. The bioavailability radar plot, which took into account the following characteristics, shows that the chosen analogues exceed the pink area zone by one parameter, indicating that their predicted oral bioavailability is good. These characteristics include flexibility, lipophilicity, saturation, size, polarity, and solubility.

SwissADME platform of 7b, 7f, 7h and 7i respectively; The colored zone (pink) was the suitable physicochemical space for oral bioavailability and lines and dots representing each compound are shown in the figure. BOILED-Egg predictive model for the compounds 7b
Experimental
Chemistry
All reactions were performed in anhydrous conditions using dry, recently distilled solvents that were obtained from SD Fine Chemicals or Sigma-Aldrich (India). Dichloromethane was distilled from CaH2 just before use, while THF was produced from salt just before use. On silica gel plates (60F-254), thin-layer chromatography (TLC) was carried out under UV light (254 and 365 nm). On silica gel, flash chromatography was carried out (230–400 mesh). A Finnigan LCQ advantage max spectrometer was used to record the ESI mass spectra and a PerkinElmer GX FTIR spectrometer was used to record the IR spectrum of these substances. In DMSO-d6, CDCl3, NMR spectra (300–400 and 100–125 MHz for 1H and 13C, respectively) were captured with TMS serving as the internal standard. The following information is recorded for 1H NMR: Hertz calculates the coupling constant J, chemical shift (ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet); and m, multiplet (dd, doublet of doublet) (Hz).
Synthesis of (2S,4R)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-hydroxy pyrrolidine-2-carboxylate 3
(2S,4R)-methyl-4-hydroxypyrrolidine-2-carboxylate hydrochloride 2 (3.0 g, 16.7 mmol) was suspended in a mixture of pyridine (25 mL) and triethylamine (4.3 mL, 30.5 mmol). The resulting mass was stirred for 20 min at ambient temperature and filtered. The filtrate was cooled to − 5 °C, then a solution of 2,4-difluoro-3-methoxybenzoyl chloride 1 (3.0 g, 14.5 mmol) in dichloromethane DCM (15 mL) was added dropwise under nitrogen atmosphere. The reaction mass was stirred for 4 h at ambient temperature. After completion of the reaction (TLC), it was filtered and the filtrate was concentrated to dryness under reduced pressure. The residue was purified by silica gel chromatography using petroleum ether-ethyl acetate (8:2) as eluent to provide 3 (3.44 g, 75%) as pale yellow solid; mp: 75–77 °C.
Synthesis of (2S,4R)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-((methylsulfonyl)oxy) pyrrolidine-2-carboxylate 4
(2S,4R)-Methyl 1-(2,4-difluoro-3-methoxybenzoyl)-4-hydroxypyrrolidine-2-carboxylate 3 (3.00 g, 9.51 mmol) was taken in DCM (15 mL) and triethylamine (3.3 mL, 23.7 mmol) was added at 0 °C under nitrogen atmosphere. Then a solution of p-tolylsulfonyl chloride (3.02 g, 16 mmol) in DCM (10 mL) was added dropwise and the resulting mixture was stirred at room temperature overnight. The reaction mixture was diluted with dichloromethane and washed with 10% aq. HCl (10 mL), saturated NaHCO3 solution (10 mL), water (10 mL) followed by brine (15 mL) and dried over anhydrous sodium sulfate. The organic layer was separated and evaporated to obtain a light yellow oil, which was purified by flash chromatography over silica gel with petroleum ether-acetone (1:1) to provide 4 (3.66 g, 82%) as a white solid; mp: 65–67 °C.
Synthesis of (2S,4S)-methyl-4-azido-1-(2,4-difluoro-3-methoxybenzoyl)pyrrolidine-2-carboxylate (5)
(2S,4R)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-((methoxy sulfonyl)oxy)pyrrolidine-2-carboxylate 4 (3.0 g, 6.39 mmol) was taken in anhydrous DMF (15 mL) under nitrogen atmosphere and sodium azide (5.2 g, 8.0 mmol) was added to the reaction mixture and stirred at 55 °C for overnight. After completion of the reaction, the reaction mass was partitioned between water (30 mL) and EtOAc (25 mL). After separation of the layers, the organic phase was washed with water followed by 0.1 M HCl (25 mL). The organic layer was then washed with brine, dried over MgSO4, separated, and evaporated to dryness under reduced pressure. The resulting oily residue was chromatographed with hexane–EtOAc (3:1) to provide 5 (1.82 g, 84%) as a yellow oil, which solidified upon standing; mp: 145–147 °C.
4.5 General procedure for the synthesis of (2S,4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-substitutedphenyl-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7)
A series of 1-phenyl-5-(prop-2-yn-1-ylthio)-1H-tetrazoles 6a–i were prepared starting from phenyl isothiocyanate which was reacted with sodium azide to give 1-phenyl-1H-tetrazole-5-thiol. Then the solution of 1-phenyl-1H-tetrazole-5-thiol (2.1 mmol), propargyl bromide (1.2 mmol), and tetrabutylammonium bromide in a mixture of triethylamine (4 mL) and DCM (6 mL) was stirred at ambient temperature for 4 h. After completion of the reaction (TLC), the reaction mixture was poured into ice-cold water (15 mL), and the solid product was filtered off, dried, and purified by column chromatography using ethyl acetate–hexane (2:8) (87%). A mixture of tetrazole 6 (1.2 mmol), pyrrolidine azide 5 (1.2 mmol), copper sulfate, and L-sodium ascorbate in DMF-H2O (10 mL, 3:2 ratio) was stirred at room temperature for 4 h. After completion of the reaction, the mixture was poured into ice water (30 mL) and extracted with 2 X 25 mL of ethyl acetate. The combined organic extract was washed twice with a saturated ammonium chloride (20 mL) and with brine (20 mL), dried over Na2SO4, and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel using n-hexane: ethyl acetate (7:3) as eluent.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(2,4-difluorophenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7a)
White solid; yield 79.8%;mp: 165–167 °C. 1H NMR (DMSO-d6, 400 MHz):δ 0.93 (1H, m, CH2), 1.16 (1H, m, CH2), 1.65 (1H, m, CH), 2.81 (3H, s, OCH3), 3.31 (2H, d, J = 8.4 Hz, CH2), 3.72 (3H, s, OCH3), 2.65 (1H, t, J = 7.8 Hz, CH), 4.88 (2H, s, SCH2), 6.81 (1H, d, J = 7.2 Hz, ArH), 7.37 (1H, dd, J = 7.2, 3.0 Hz, ArH), 7.54 (1H, d, J = 7.2 Hz, ArH), 8.03 (1H, dd, J = 7.2, 3.0 Hz, ArH), 8.44 (1H, d, J = 7.4 Hz, ArH), 8.62 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 24.02, 34.90, 48.56, 51.79, 55.27, 61.98, 62.28, 102.47, 119.78, 123.59, 125.66, 128.26, 128.40, 135.78, 142.35, 150.03, 151.48, 156.23, 160.06, 161.41, 172.03, 176.17. IR (KBr, cm−1): 3037.75 (= CH), 2927.23, 2908.34 (CH), 1699.86 (ester C=O), 1637.56 (amide C=O), 1597.22, 1514.65 (Ar–C=C), 1406.41 (C=N), 1368.12 (N=N), 1209.47 (C–S–C), 1103.49 (C–O–C), 1040.23 (C–F);m/z: 593 [M + H]+.
(2S,4S)-methyl-4-(4-(((1-(2-chloro-4-fluorophenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-1-(2,4-difluoro-3-methoxybenzoyl)pyrrolidine-2-carboxylate (7b)
White crystalline solid; yield 76.1%;mp: 160–162 °C.1H NMR (DMSO-d6, 400 MHz):δ 1.57 (1H, m, CH2), 1.79 (1H, t, J = 7.8 Hz, CH2), 2.14 (1H, m, CH), 2.91 (2H, d, J = 8.0 Hz, CH2), 3.23 (3H, s, OCH3), 3.43 (3H, s, OCH3), 3.67 (1H, t, J = 7.8 Hz, CH), 4.77 (2H, s, SCH2), 7.24 (1H, d, J = 7.3 Hz, ArH), 7.75 (1H, dd, J = 7.4, 3.2 Hz, ArH), 7.79 (1H, d, J = 7.3 Hz, ArH), 7.85 (1H, dd, J = 7.4, 3.2 Hz, ArH), 8.79 (1H, d, J = 7.3 Hz, ArH), 9.46 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 24.71, 35.41, 40.86, 54.98, 57.98, 61.03, 62.50, 102.56, 113.34, 126.03, 128.40, 128.78, 135.68, 139.82, 147.92, 150.19, 151.32, 155.51, 165.06, 166.02, 171.20. IR (KBr, cm−1): 3042.49 (= CH), 2966.64, 2931.91 (CH), 1699.22 (ester C=O), 1672.11 (amide, C=O), 1594.50, 1539.93 (Ar–C=C), 1427.03 (C=N), 1279.37 (C–S–C), 1173.97 (C–O–C), 1104.69 (C–F), 1021.49 (C–F) m/z: 609 [M + H]+.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(2,4-dimethoxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7c)
Pale yellow oil; yield 70.2%;1H NMR (DMSO-d6, 400 MHz):δ 2.12 (1H, m, CH2), 2.27 (1H, m, CH2), 2.69 (1H, m, CH), 2.78 (1H, d, J = 7.3 Hz, CH2), 2.89 (1H, d, J = 7.3 Hz, CH2), 3.07 (6H, s, OCH3), 3.24 (3H, s, OCH3), 3.55 (3H, s, OCH3), 3.84 (1H, t, J = 7.4 Hz, CH), 4.95 (2H, s, SCH2), 7.03 (1H, s, ArH), 7.24 (1H, d, J = 7.1 Hz, ArH), 7.41 (1H, t, J = 7.4 Hz, ArH), 7.86 (1H, d, J = 7.1 Hz, ArH), 8.23 (2H, d, J = 7.4 Hz, ArH), 8.83 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 23.81, 36.62, 46.20, 51.27, 56.31, 59.60, 62.37, 64.83, 109.37, 113.21, 116.39, 124.38, 127.64, 128.02, 131.58, 137.22, 146.85, 154.39, 157.45, 158.88, 172.63, 174.14. IR (KBr, cm−1): 3051.26 (= CH), 2935.33, 2875.15 (CH), 1706.45 (ester C=O), 1653.91 (amide C=O), 1585.62, 1524.29 (Ar–C=C), 1422.47 (C=N), 1215.10 (C–S–C), 1166.01 (C–O–C), 1034.77 (C–F). m/z: 617 [M + H]+.
(2S,4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(p-tolyl)-1H-tetrazol-5-yl) thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7d)
Colourless gummy liquid; yield: 68%;1H NMR (DMSO-d6, 400 MHz):δ 1.49 (1H, m, CH2), 1.78 (1H, m, CH), 2.20 (3H, s, CH3), 2.57 (1H, dd, J = 7.4, 2.7 Hz, CH), 3.37 (3H, s, OCH3), 3.62 (1H, d, J = 7.5 Hz, CH2), 3.70 (1H, d, J = 7.5 Hz, CH2), 4.03 (3H, s, OCH3), 4.37 (1H, t, J = 7.8 Hz, CH), 4.90 (2H, s, SCH2), 7.33 (1H, d, J = 7.4 Hz, ArH), 7.72 (2H, d, J = 7.3 Hz, ArH), 8.81 (2H, d, J = 7.3 Hz, ArH), 8.87 (1H, dd, J = 7.4, 3.1 Hz, ArH), 9.37 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 22.92, 29.10, 35.27, 48.79, 51.56, 56.54, 62.22, 63.50, 104.01, 118.12, 119.28, 121.43, 122.73, 124.06, 130.18, 132.77, 142.07, 151.10, 155.66, 163.22, 171.45, 172.38. IR (KBr, cm−1): 3032.59 (= CH), 2956.22, 2896.71 (CH), 1713.31(ester C=O),1671.06 (amide C=O), 1551.05, 1472.97 (Ar–C=C), 1408.22 (C=N), 1323.46 (N=N), 1258.64 (C–S–C), 1171.07 (C–O–C), 1026.60 (C–F), 992.61 (C–F) m/z: 571 [M + H]+.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(4-nitrophenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7e)
Yellow crystalline solid; yield 92%;mp: 145–147 °C.1H NMR (DMSO-d6, 400 MHz):δ 2.06 (1H, m, CH2), 2.25 (1H, m, CH2), 2.74 (1H, m, CH), 2.71 (1H, d, J = 7.4 Hz, CH2), 2.79 (1H, d, J = 7.4 Hz, CH2), 3.26 (3H, s, OCH3), 3.58 (3H, s, OCH3), 3.79 (1H, t, J = 7.8 Hz, CH), 4.62 (2H, s, SCH2), 7.01 (1H, d, J = 7.0 Hz, ArH), 7.36 (1H, t, J = 7.5 Hz, ArH), 7.84 (1H, s, triazole H), 8.21 (2H, d, J = 7.4 Hz, ArH), 8.51 (2H, d, J = 7.4 Hz, ArH). 13C NMR (DMSO-d6, 100 MHz):δ 22.97, 25.79, 37.79, 49.81, 51.36, 58.83, 60.75, 114.28, 114.47, 118.71, 125.56, 127.76, 128.02, 130.85, 137.66, 142.35, 145.58, 157.36, 158.50, 172.68, 176.13. IR (KBr, cm−1): 3022.99 (= CH), 2927.05, (CH), 1730.56 (ester C=O), 1640.15 (amide C=O), 1601.39, 1512.78 (Ar–C=C), 1429.07 (C=N), 1336.73 (N=N), 1261.23 (C–S–C), 1135.37 (C–O–C), 1030.78 (C–F), 983.11 (CF) m/z: 602 [M + H]+.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(2-nitrophenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7f)
Yellow crystalline solid, yield: 85%, mp: 132–134 °C.1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.03 (1H, m, CH2), 2.21 (1H, m, CH2), 2.73 (1H, m, CH), 2.78 (1H, d, J = 7.3 Hz, CH2), 2.96 (1H, d, J = 7.3 Hz, CH2), 3.21 (3H, s, OCH3), 3.45 (3H, s, OCH3), 3.80 (1H, t, J = 7.7 Hz, CH), 4.68 (2H, s, SCH2), 7.06 (1H, d, J = 6.8 Hz, ArH), 7.30 (1H, t, J = 7.7 Hz, ArH), 7.73 (1H, dd, J = 7.5, 3.2 Hz, ArH), 7.88 (1H, dd, J = 7.5, 3.1 Hz, ArH), 8.05 (2H, m, ArH), 8.50 (1H, s, triazole H).13C NMR (DMSO-d6, 100 MHz):δ 24.13, 26.37, 35.60, 48.44, 50.37, 57.66, 61.91, 114.17, 115.33, 118.45, 124.88, 127.30, 128.21, 130.92, 135.55, 142.37, 145.68, 156.69, 157.11, 171.37, 173.94. IR (KBr, cm−1): 3081.31 (= CH), 2934.79, 2834.30 (CH), 1719.60 (ester C=O), 1646.18 (amide C=O), 1578.90 (Ar–C=C), 1431.90 (C=N), 1328.11 (N=N), 1224.83 (C–S–C), 1142.66 (C–O–C), 1026.81 (C–F), 977.52 (C–F) m/z: 602 [M + H]+.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-phenyl-1H-tetrazol-5-yl) thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7 g)
Brown color solid; yield 65%;mp: 161–163 °C.1H NMR (CDCl3, 400 MHz):δ 2.20 (1H, m, CH2), 2.41 (1H, m, CH2), 2.82 (1H, d, J = 7.0 Hz, CH2), 3.09 (1H, m, CH), 3.30 (1H, d, J = 7.0 Hz, CH2), 3.55 (3H, s, OCH3), 3.83 (3H, s, OCH3), 3.96 (1H, t, J = 7.5 Hz, CH), 4.93 (2H, s, SCH2), 6.83 (1H, d, J = 7.2 Hz, ArH), 7.12 (3H, m, ArH), 7.28 (1H, d, J = 7.2 Hz, ArH), 7.62 (1H, t, J = 7.4 Hz, ArH), 7.94 (1H, d, J = 7.2 Hz, ArH), 8.77 (1H, s, triazole H). 13C NMR (CDCl3, 100 MHz):δ23.21, 35.60, 43.67, 52.75, 55.33, 62.50, 63.97, 116.33, 118.43, 122.80, 124.55, 127.28, 135.61, 148.67, 152.34, 155.81, 157.33, 161.73, 170.39, 173.61. IR (KBr, cm−1): 3043.11 (= CH), 2931.37, (CH), 1727.34 (ester C=O), 1638.91 (C = O), 1606.46, 1534.24 (Ar–C=C), 1416.27 (C=N), 1343.44 (N=N), 1254.37 (C–S–C), 1144.27 (C–O–C), 1025.77 (C–F), 979.24 (C–F) m/z: 557 [M + H]+.
4.13 (2S, 4S)-Methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(2-hydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7 h) Colourless gummy liquid; yield 81%;1H NMR (DMSO-d6, 400 MHz):δ 2.16 (1H, m, CH2), 2.31 (1H, m, CH2), 2.76 (1H, m, CH), 3.13 (1H, d, J = 7.1 Hz, CH2), 3.25 (1H, d, J = 7.1 Hz, CH2), 3.50 (3H, s, OCH3), 3.71 (3H, s, OCH3), 3.89 (1H, t, J = 7.4 Hz, CH), 5.10 (2H, s, SCH2), 5.67 (1H, brs, OH), 6.86 (1H, d, J = 7.0 Hz, ArH), 7.06 (1H, dd, J = 7.8, 3.1 Hz, ArH), 7.20 (1H, dd, J = 7.8, 3.1 Hz, ArH), 7.45 (1H, d, J = 7.0 Hz, ArH), 7.64 (1H, t, J = 7.5 Hz, ArH), 7.94 (1H, d, J = 7.0 Hz, ArH), 8.86 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 22.31, 36.20, 44.67, 52.66, 56.45, 61.56, 63.41, 106.70, 118.72, 119.94, 123.85, 124.61, 127.11, 135.62, 148.91, 153.44, 154.26, 156.70, 157.92, 161.46, 171.04, 173.37. IR (KBr, cm−1): 3458.24 (OH), 3084.34 (= CH), 2963.55, 2874.23 (CH), 1716.94 (ester C = O), 1661.18 (amide C=O), 1555.60 (Ar–C=C), 1428.63 (C=N), 1320.83 (N=N), 1204.77 (C–S–C), 1166.18 (C–O–C), 1020.61 (C–F) m/z: 573 [M + H]+.
(2S, 4S)-methyl-1-(2,4-difluoro-3-methoxybenzoyl)-4-(4-(((1-(2,4-dihydroxyphenyl)-1H-tetrazol-5-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboxylate (7i)
Pale yellow solid; yield 88%;mp: 128–130 °C.1H NMR (DMSO-d6, 400 MHz):δ 1.48 (1H, m, CH2), 1.62 (1H, m, CH2), 2.07 (1H, m, CH), 2.72 (1H, d, J = 7.2 Hz, CH2), 2.81 (1H, d, J = 7.2 Hz, CH2), 2.97 (3H, s, OCH3), 3.52 (3H, s, OCH3), 3.66 (1H, t, J = 7.5 Hz, CH), 4.90 (2H, s, SCH2), 5.50 (2H, brs, OH), 6.58 (1H, s, ArH), 6.75 (1H, d, J = 7.0 Hz, ArH), 7.21 (1H, t, J = 7.4 Hz, ArH), 7.42 (1H, d, J = 7.0 Hz, ArH), 7.69 (1H, d, J = 7.0 Hz, ArH), 8.21 (1H, s, triazole H). 13C NMR (DMSO-d6, 100 MHz):δ 25.16, 37.26, 41.70, 51.79, 56.69, 60.50, 65.27, 105.76, 117.02, 119.78, 123.59, 128.45, 135.78, 141.54, 148.82, 153.45, 154.38, 156.87, 157.56, 161.46, 172.80, 175.67. IR (KBr, cm−1): 3486.30 (OH), 3055.09 (= CH), 2982.59, 2896.08 (CH), 1712.63 (ester C=O), 1668.10 (ester C=O), 1598.62, 1552.60 (Ar–C=C), 1467.59 (C=N), 1325.73 (N=N), 1256.93 (C–S–C), 1173.20 (C–O–C), 1103.32 (C–F), 1026.40 (C–F) m/z: 589 [M + H]+.
*General experimental details of biological activity evaluation, computational docking studies and copies of 1H NMR, 13C NMR, and IR spectrums are included in supporting information.*
Conclusion
New series of 1,2,3-triazoles & tetrazoles incorporated pyrrolidine derivatives have been prepared and evaluated for their anticancer activity. In the current study, novel pyrrolidine coupled to dual heterocycles was conveniently synthesised, and all the title compounds were examined using 1H NMR, 13C NMR, IR, and mass spectrometry. The results of this study demonstrate that the compounds7a, 7b, 7e, 7f, 7h, and 7i have a promising anticancer activity against Hela (human cervical epithelioid carcinoma), MCF-7 (human breast adenocarcinoma), HCT-116 (human colon carcinoma), and HepG2 cells (human caucasian hepatocyte carcinoma). In particular, the prepared scaffolds 7b and 7i, showed remarkable anticancer activity against HCT-116 cancer cell line with IC50values 1.48 ± 0.08 μM, 1.38 ± 0.06 μM respectively. Molecular docking simulations of targeting the active site of human breast and colon carcinoma proteins (3HB4, 6IBZ) were used in addition to the in vitro analysis to explore potential interactions of these analogues with the receptor. On the other hand, the docking results also correlate well with the biological activity and compounds with potent inhibitory activities towards anticancer growths were further evaluated for their ADMET and physicochemical properties. Upon keen observation of the obtained results, it can be concluded that these molecules become lead molecules for further synthetic and biological evaluation.
Data availability
Not Applicable.
Abbreviations
- CYP2C19:
-
Cytochrome P450 2C19 gene family
- LGA:
-
Lamarckian Genetic Algorithm
- ADMET:
-
Absorption, distribution, metabolism, excretion and toxicology
- TPSA:
-
Topological polar surface area
- ADT:
-
Auto dock tools
- PDBQT:
-
Protein Data Bank includes partial charges ('Q') and atom types ('T')
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DMJ (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127:2893–2917. https://doi.org/10.1002/ijc.25516&GLOBOCAN2020:NewGlobalCancerData
Gottesman MM (2002) Mechanisms of cancer drug resistance. Annu Rev Med 53:615–627. https://doi.org/10.1146/annurev.med.53.082901.103929
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Burdall SA, Speirs MR (2003) Spearseast cancer cell lines: friend or foe. Breast Cancer Res 5:89–95. https://doi.org/10.1186/bcr577
Sweeney EE, Mcdaniel RE, Maximov PY, Fan P, Craig V (2013) Models and mechanisms of acquired antihormone resistance in breast cancer: significant clinical progress despite limitations. Horm Mol Biol Clin Investig 9:143–163. https://doi.org/10.1515/hmbci-2011-0004
Zur Hausen H (1989) Papillomaviruses in anogenital cancer as a model to understand the role of viruses in human cancers. Cancer Res 49:4677–4681
Wani MY, Bhat AR, Azam A, Choi I, Athar F (2012) Probing the antiamoebic and cytotoxicity potency of novel tetrazole and triazine derivatives. Eur J Med Chem 48:313–320. https://doi.org/10.1016/j.ejmech.2011.12.033
OrnellaM AM, Mariateresa G, TraceyP ValentinaB, Antonio C, Pierluigi C, Giovanni S, EttoreN AAG, Gian CT (2011) Replacement of the double bond of antitubulin chalcones with triazoles and tetrazoles: Synthesis and biological evaluation. Bioorg Med Chem Lett 21:764–768. https://doi.org/10.1016/j.bmcl.2010.11.113
Sabbah M, Fontaine F, Grand L, Boukraa M, Efrit ML, Doutheau A, Soulre L, Queneau Y (2012) Synthesis and biological evaluation of new N-acyl-homoserine-lactone analogues, based on triazole and tetrazole scaffolds, acting as LuxR-dependent quorum sensing modulators. Bioorg Med Chem 20:4727–4736. https://doi.org/10.1016/j.bmc.2012.06.007
Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A, Karnik SS (1995) Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. J Biol Chem 270:2284–2289. https://doi.org/10.1074/jbc.270.5.2284
El-Sayed WA, Abdel Megeid RE, Abbas HA (2011) Synthesis and antimicrobial activity of new 1-[(tetrazol-5-yl)methyl]indole derivatives, their 1,2,4-triazole thioglycosides and acyclic analogs. Arch Pharm Res 34:1085–1096. https://doi.org/10.1007/s12272-011-0706-y
Upadhayaya RS, Sinha N, Jain S, Kishore N, Chandra R, Arora S (2004) Optically active antifungal azoles: synthesis and antifungal activity of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-{2-[4-aryl-piperazin-1-yl]-ethyl}-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg Med Chem 12:2225–2238. https://doi.org/10.1016/j.bmc.2004.02.014
Dougherty AM, Guo H, Westby G, LiuY SE, Guo J, Mehta A, Norton P, Gu B, Block T (2007) A substituted tetrahydro-tetrazolo-pyrimidine is a specific and novel inhibitor of hepatitis b virus surface antigen secretion. Antimicrob Agents Chemother 51:4427–4437. https://doi.org/10.1128/aac.00541-07
Gundugola AS, Chandra KL, Perchellet EM, Waters AM, Perchellet JPH, Rayat S (2010) Synthesis and antiproliferative evaluation of 5-oxo and 5-thio derivatives of 1,4-diaryl tetrazoles. Bioorg Med Chem Lett 20:3920–3924. https://doi.org/10.1016/j.bmcl.2010.05.012
Wang SQ, Wang YF, Xu Z (2019) Tetrazole hybrids and their antifungal activities. Eur J Med Chem 170:225–234. https://doi.org/10.1016/j.ejmech.2019.03.023
Gao C, Chang L, Xu Z, Yan XF, Ding C, Zhao F, Wu X, Feng LS (2018) Recent advances of tetrazole derivatives as potential anti-tubercular and anti-malarial agents. Eur J Med Chem 163:404–412. https://doi.org/10.1016/j.ejmech.2018.12.001
Purohit P, Pandey AK, Singh D, Chouhan PS, Ramalingam KM, Goyal N, Lal J, Chauhan PM (2017) An insight into tetrahydro-β–tetrazole hybrids: synthesis and bio evaluation as potent anti-leishmanial agents. Med Chem Commun 8:1824–1834. https://doi.org/10.1039/C7MD00125H
Kaushik N, Kumar N, Kumar A (2016) Synthesis, antioxidant and antidiabetic activity of 1-[(5-Substituted phenyl)-4,5-dihydro-1H-pyrazol-3-yl]-5-phenyl-1H-tetrazole. Indian J Sci 78:352–359. https://doi.org/10.4172/pharmaceutical-sciences.1000125
McGuire JJ, Russell CA, Bolanowska WE, Freitag CM, Jones CS, Kalman TI (1990) Biochemical and growth inhibition studies of methotrexate and aminopterin analogues containing a tetrazole ring in place of the γ-carboxyl group. Cancer Res 50:1726–1731
Song MX, Deng XQ (2018) Recent developments on triazole nucleus in anticonvulsant compounds: a review. J Enzyme Inhib Med Chem 33:453–478. https://doi.org/10.1080/14756366.2017.1423068
Jordão AK, Ferreira VF, Souza TML, De Souza Faria GG, MacHado V, Abrantes JL, De Souza MCBV, Cunha AC (2011) Synthesis and anti-HSV1 activity of new 1,2,3-triazole derivatives. Bioorganic Med Chem 19:1860–1865. https://doi.org/10.1016/j.bmc.2011.02.007
BoechatN FerreiraVF, FerreiraSB FMDLG, Da Silva FDC, Bastos MM, Costa MDS, Lourenço MCS, Pinto AC, Krettli AU, Aguiar AC, Teixeira BM, Da Silva NV, Martins PRC, Bezerra FAFM, Camilo ALS, Da Silva GP, Costa CCP (2011) Novel 1,2,3-triazole derivatives for use against mycobacterium tuberculosis H37Rv (ATCC 27294) strain. J Med Chem 54:5988–5999. https://doi.org/10.1021/jm2003624
Mady MF, Awad GEA, Jørgensen KB (2014) Ultrasound-assisted synthesis of novel 1,2,3-triazoles coupled diaryl sulfone moieties by the CuAAC reaction, and biological evaluation of them as antioxidant and antimicrobial agents. Eur J Med Chem 84:433–443. https://doi.org/10.1016/j.ejmech.2014.07.042
Garudachari B, Isloor AM, Satyanarayana MN, Fun HK, Hegde G (2014) Click chemistry approach: regioselective one-pot synthesis of some new 8trifluoromethylquinoline based 1,2,3-triazoles as potent antimicrobial agents. Eur J Med Chem 74:324–332. https://doi.org/10.1016/j.ejmech.2014.01.008
Bollu R, Palem JD, Bantu R, Guguloth V, Nagarapu L, Polepalli S, Jain N (2014) Rational design, synthesis and anti-proliferative evaluation of novel 1,4-benzoxazine-[1,2,3]triazole hybrids. Eur J Med Chem 89:138–146. https://doi.org/10.1016/j.ejmech.2014.10.051
Suresh M, Kumar AS, Gorle S, Singh M, Lavanya P, Jonnalagadda SB (2013) Synthesis and antioxidant activity of 1,2,4-triazole linked thieno[2,3-d]pyrimidine derivatives. Lett Drug Des Discov 10:186–193. https://doi.org/10.2174/157018013804725152
Kharb R, Sharma PC, Yar MS (2011) Pharmacological significance of triazole scaffold. J Enzy Inhib Med Chem 26:1–21. https://doi.org/10.3109/14756360903524304
Milton NG (2001) Inhibition of catalase activity with 3-amino-triazole enhances the cytotoxicity of the Alzheimer’s amyloid-beta peptide. Neurotoxicology 22:767–774. https://doi.org/10.1016/S0161-813X(01)00064-X
Varvaresou A, Siatra-Papastaikoudi T, Tsotinis A, Tsantili-Kakoulidou A, Vamvakides A (1998) Synthesis, lipophilicity and biological evaluation of indole-containing derivatives of 1,3,4-thiadiazole and 1,2,4-triazole. Farmaco 53:320–326. https://doi.org/10.1016/S0014-827X(98)00024-X
Turan-Zitouni G, Kaplancikli ZA, Ozdemir A, Chevallet P, Kandilci HB, Gumusel B (2007) Studies on 1,2,4-triazole derivatives as potential anti-inflammatory agents. Arch Pharm (Weinheim) 340:586–590. https://doi.org/10.1002/ardp.200700134
Berk B, Aktay G, Yesilada E, Ertan M (2001) Synthesis and pharmacological activities of some new 2-[1-(6-methoxy-2-naphthyl)ethyl]-6-(substituted)benzylidene thiazolo[3,2-b]-1,2,4-triazole-5(6H)-one derivatives. Pharmazie 56:613–616. https://doi.org/10.1002/chin.200147107
Zhan P, Chen X, Li X, Li D, Tian Y, Chen W, Pannecouque C, Clercq E, Liu X (2011) Design, synthesis and biological evaluation of Novel 2-(2-(2,4-Dichloro phenyl)-2H-1,2,4-triazol-3-ylthio)-N-arylacetamides As Potent HIV-1 inhibitors. Eur J Med Chem 46:5039–5045. https://doi.org/10.1016/j.ejmech.2011.08.011
Li YL, Xu WF (2004) Design, synthesis, and activity of caffeoyl pyrrolidine derivatives as potential gelatinase inhibitors. Bioorg Med Chem 12:5171–5180. https://doi.org/10.1016/j.bmc.2004.07.025
LiX LY, Xu WF (2006) Design, synthesis, and evaluation of novel galloyl pyrrolidine derivatives as potential anti-tumor agents. Bioorg Med Chem 14:1287–1293. https://doi.org/10.1016/j.bmc.2005.09.031
Thotla K, Noolea VG, Kedikaa B, Krishna Reddy CH (2020) Synthesis of 5-{[(1-Aryl-1H-1,2,3-triazol-4-yl)methyl]sulfanyl}-1-phenyl-1H-tetrazoles. Russian J Org Chem 56:1077–1081. https://doi.org/10.1134/S1070428020060172
Riss TL, Moravec RA, Niles AL, Duellman S, Benink HA, Worzella TJ, Minor L (2016) Cell Viability Assays, Eli Lilly & Company and the National Center for Advancing Translational Sciences. https://www.ncbi.nlm.nih.gov/books/NBK144065/pdf/Bookshelf_NBK144065.pdf
Bose DS, Idrees M, Jakka NM, Rao JV (2010) Diversity-oriented synthesis of quinolines via Friedlander annulation reaction under mild catalytic conditions. J Comb Chem 12:100–110. https://doi.org/10.1021/cc900129t
Abu Bakar MF, Maryati M, Rahmat A, Burr SA, Fry JR (2010) Cytotoxicity, cell cycle arrest, and apoptosis in breast cancer cell lines exposed to an extract of the seed kernel of Mangifera pajang (bambangan). Food Chem Toxicol 48:1688–1697. https://doi.org/10.1016/j.fct.2010.03.046
Mazumdar M, Fournier D, Zhu DW, Cadot C, Poirier D, Lin SX (2009) Binary and ternary crystal structure analyses of a novel inhibitor with 17β-HSD type 1: a lead compound for breast cancer therapy. Biochem J 424:357–366. https://doi.org/10.1042/BJ20091020
Boutard N, BialasA SA, Guzik P, Banaszak K, Biela A, Bien M, Buda A, Bugaj B, Cieluch E, Cierpich A, Dudek L, Eggenweiler HM, Fogt J, Gaik M, Gondola A, Jakubiec K, Jurzak M, Kitlinska A, Kowalczyk P, Kujawa M, KwiecinskaK LM, Lindemann R, Maciuszek M, Mikulski M, Niedziejko P, Obara A, Pawlik H, Rzymski T, Sieprawska-Lupa M, Sowinska M, Szeremeta-Spisak J, Stachowicz A, Tomczyk MM, Wiklik K, Wloszczak L, Ziemianska S, Zarebski A, Brzozka K, Nowak M, Fabritius CH (2019) Synthesis of amide and sulfonamide substituted N-aryl 6-aminoquinoxalines as PFKFB3 inhibitors with improved physicochemical properties. Bioorg Med Chem Lett 29:646–653. https://doi.org/10.1016/j.bmcl.2018.12.034
ACD/ChemSketch, version 2020.2.1 (2021) Advanced Chemistry Development, Inc., Toronto, www.acdlabs.com.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Chem Inform 3:33. https://doi.org/10.1186/1758-2946-3-33
Askowski RA, Jabłońska J, Pravda L, Varekova RS, Thornton JM (2018) PDBsum: structural summaries of PDB entries. Protein Sci 27:129–134. https://doi.org/10.1002/pro.3289
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 16:2785–2791. https://doi.org/10.1002/jcc.21256
Antoine D, Olivier M, Vincent Z (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Bakchi B, Dileep Krishna A, Sreecharan E, Jaya Ganesh VB, Niharika M, Maharshi S, Srinivasa Babu P, Dilep Kumar S, Richie RB, Afzal BS (2022) An overview on applications of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: a medicinal chemist’s perspective. J Mol Struct 1259:132712. https://doi.org/10.1016/j.molstruc.2022.132712
Antoine D, Olivier M, Vincent Z (2014) iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model 54:3284–3301. https://doi.org/10.1021/ci500467k
Acknowledgements
The authors are thankful to Department of Chemistry, GITAM School of Science, GITAM (Deemed to be University) for providing required facilities for completion of the research work. We also extend our gratitude towards Jawaharlal Nehru Technological University Hyderabad for providing biological activities and Osmania University, Hyderabad for providing Molecular Modelling studies.
Funding
None.
Author information
Authors and Affiliations
Contributions
SKG, AAK, AJ in-charge of synthesis, write up and characterization studies; TRA and KC write up the computational molecular docking calculations and biological activities.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gandham, S.K., Kudale, A.A., Allaka, T.R. et al. New tetrazolopyrrolidine-1,2,3-triazole analogues as potent anticancer agents: design, synthesis and molecular docking studies. Mol Divers (2023). https://doi.org/10.1007/s11030-023-10762-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11030-023-10762-z