
Abstract
In an attempt to develop potent anti-cancer agents, a new 1,3,4-substituted-thiadiazole derivatives (8b-g), starting from 4-substituted-thiazol-2-chloroacetamides (4b-g), were synthesized and evaluated for their cytotoxic effects on multiple human cancer cell lines, including the hepatocellular carcinoma (HEPG-2), human lung carcinoma (A549), human breast carcinoma (MCF-7) and pseudo-normal human embryonic liver (L02) cancer cell lines by an MTT assay. Among all synthesized compounds, compound 8d showed the potent anti-cancer activities with GI50 values of 2.98, 2.85 and 2.53 μM against MCF-7, A549 and HepG-2 cell lines respectively as compared to standard drug Doxorubicin. Furthermore, molecular modelling studies have spotlighted the anchoring role of 1,3,4-substituted-thiadiazole moiety in bonding and hydrophobic interaction with the key amino acid residues. Therefore, these results can provide promising starting points for further development of best anti-cancer agents.
Graphical Abstract

Similar content being viewed by others

Introduction
Cancer is presently one of the main causes of death worldwide. The International Agency for Research on Cancer (IARC) estimated that globally, 1 in 5 people develop cancer during their lifetime, and 1 in 8 men and 1 in 11 women die from the disease suggesting that more than 50 million people are living within five years of a past cancer diagnosis [1]. In past decades, anticancer research on the design of effective oncology drugs for the application in chemotherapy has improved treatments which led to remarkable results and many drugs have been approved [2, 3]. However, many of the approved drugs still being characterized by high systemic toxicity mainly due to the lack of tumor selectivity and present pharmacokinetic drawbacks, including low water solubility, that negatively affect the drug circulation time and bioavailability which limit its clinical application [4]. The above stated disadvantages of conventional anticancer drugs are the reason why the development of new anticancer drug or improvement in present anticancer drug is still demanding. Therefore, the newer generations of CDK9 inhibitors are already raising as an anticancer therapy and present ongoing research in this direction are helping to develop better, more selective inhibitors.
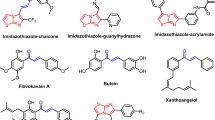
Thus, the best suitable inhibition strategies are required to be able to distinguish between normal rapidly proliferating cells like the T cells and cancer cells. Thiazole and thiadiazole possess unique properties that make them useful scaffolds in medicinal chemistry [5,6,7,8,9,10], few examples are shown in Fig. 1. As bioisosteres of pyrimidines, their derivatives can potentially interact with DNA and RNA and can readily cross cell membranes and interact with biological compounds in unique ways. For example, Filanesib (Array-520, Fig. 1) is anti-cancer drug used specifically for multiple myeloma. The best strategy showed when it was combined with bortezomib and dexamethasone had a favorable safety profile. The resistance to Dabrafenib (Fig. 1) and other BRAF inhibitors could be reduced when the Dabrafenib was combined with the MEK inhibitor Trametinib [11]. Recently Noblejas-López et al. had verified that CDK9 PROTAC THAL-SNS-032 showed potent and efficient anti-tumor properties [12]. However, no clinical trials of THAL-SNS-032 (Fig. 1, see structure SNS-032) have been initiated till date.

Some examples of thiazole and diathiazole scaffold-based (marked in pink circle) anti-cancer drugs
Literature data revealed that many research groups used these scaffolds as a basic core for the development of new molecules for anticancer activity. In this study, we have followed the same strategy and proposed to synthesize novel derivatives (8b-g) having two basic scaffolds, on one side is thiazole ring and on another side is 5-aminodiathiazole moiety that could mimic Dabrafenib’s side chain (2-aminopyrimidine) to some extent (Fig. 2). Additionally, hydrophobic side chains on these scaffolds could offer the hydrophobic sphere in a molecule which could contribute to enhance the CDK9 selectivity.

Structural modifications of 4-substituted-thiazol-2-chloroacetamides (4a-g) clubbed with 5-amino-1,3,4-thiadiazole-2-thiol (7) moiety
Results and discussion
To confirm the anticancer properties for our new compounds (8b-g), first we have referred our previously reported compounds (4a-g) [13], and interestingly, the docking studies of them confirmed their potential affinity to selective CDK9/Cyclin T1 (1BLQ), inhibitors (Fig. 4a and Table 2). Based on this results; we proposed the design and synthesis of 1,3,4-substituted-thiadiazole compounds (8a-g) as shown in Fig. 2. Although, Gang Yan et al. has reported the compound 8a, however its anticancer activity is not yet fully explored [14]. The structures of the newly synthesized compounds were elucidated by FT-IR, 1H NMR, 13C NMR, and LC − MS analysis methods. These compounds (8a-g) were screened for in vitro anticancer activity against hepatocellular carcinoma cell lines (HEPG-2), human lung carcinoma cell line (A549) and human breast carcinoma cell (MCF-7) and pseudo-normal human embryonic live cell line (L02) by MTT assay. In addition, molecular docking studies were applied to investigate the anti-proliferative effects of these novel compounds.
Synthetic chemistry
First we have synthesized 4-substituted-thiazol-2-chloroacetamide compounds (4a-m) starting from aromatic ketones and ethyl acetoacetates derivatives in three steps using synthetic methods reported in our previous research articles (Scheme 1) [13] and used them for the synthesis of 1,3,4-substituted-diathiazole derivatives (8a-g). The synthetic protocols for (4a-m) and (8a-g) are given in the Additional file 1 (see Additional file 1 for general experimental procedure).

Synthesis of 4-substituted-thiazol-2-chloroacetamide compounds (4a-m); Reagents and conditions: a. Bromination: Br2/AcOH, 0–5 °C, 3 h, (for 2k: Bromination: Br2/CHCl3, 55 °C, 4 h); b Hantzsch reaction: thiourea, EtOH, reflux, 8h; c Amidation: CAC, TEA, DMF, 0–5 °C, 0.5–2.5 h
The intermediates 2-aminothiazoles derivatives (3a-m) were prepared by reaction of aromatic ketones and Br2 in acetic acid or chloroform followed by Hantzsch reaction of direct condensation of intermediates (2a-m) with thiourea in a good to excellent yields. Further amidation reaction using -CAC (chloro acetyl chloride) in DMF furnished 4-substituted-thiazol-2-chloroacetamide derivatives (4a-m) in excellent yield (Scheme 1). We have followed reported procedure and optimized reaction conditions to obtain these compounds in better yield [13, 15,16,17,18,19].
Our main focus was to synthesize 1,3,4-substituted-thiadiazoles derivatives (8a-g). The synthetic strategy has been developed by clubbing of the 4-substituted-thiazol-2-chloroacetamide compounds (4a-g) with 5-amino-1,3,4-thiadiazole-2-thiol (7) via thial linkage to furnish thiadiazole derivatives (8a-g). The optimized reaction condition for (8a-g) is depicted in Scheme 2. The excellent yield of these derivatives (8a-g) was achieved by substitution reaction of compounds (4a-g) with 5-amino-1,3,4-thiadiazole-2-thiol (7) in THF at room temperature (Scheme 2, 78–24%). Surprisingly, the substitution reaction worked well for any substituent present on thiazole ring such as -Ph, -4-Br-Ph,-4-Cl-Ph, -4-OMe-Ph, -4-Me-Ph, -4-NO2-Ph and -4-F-Ph (Scheme 2, 8a-g).

Synthesis of substituted-thiadiazole derivatives (8a-g); Reagents and conditions: a. NaHCO3, EtOH, reflux, 5 h, 90%; b. K2CO3, THF, rt, 0.5–1 h, 78–94%
The 8d was synthesized with less yield (8d:78%), this may be due to the strong positive resonance effect of -OMe group (R2) present on the phenyl ring. Especially, when -4-Br-Ph, -4- Cl-Ph, -NO2-Ph and -4-F-Ph groups were present on thiazole ring then highest yields observed (Scheme 2, 8b, 8c, 8f and 8 g: 91–94%). This may be due to the negative resonance effect of -NO2 group and strong electro-negative atoms such as -Br, -Cl, and -F which made electrophilic substitution reaction better for the 4-substituted-thiazol-2-chloroacetamide compounds 4a to 4 g.
Anti-proliferative activity
Several structural alterations in compounds (8a-g) in relation to the substitution at C4 of the thiazole moiety were made to examine whether the nature of the substituent would affect selective toxicity for hepatocellular carcinoma cell lines (HEPG-2), human lung carcinoma cell line (A549) and human breast carcinoma cell line (MCF-7) and pseudo-normal human embryonic liver cell line (L02) by MTT assay. The doxorubicin is used as a standard drug; it is a reasonable standard cytotoxic agent that is a well-known chemotherapy medication. Interestingly, the compounds 4c, 4j, 8b, 8d and 8f showed the best anti-proliferative activity with IC50 values in the range of 1.82 to 4.07 µM against the MCF-7 cell line. They were also equally potent against A549 and HepG-2 cell lines (GI50 values in the range of 2.61 to 18.43 and 2.38 to 5.86 µM respectively) as compared to the doxorubicin (GI50 = 3.26 and 3.87 µM respectively, Table 1).
As hypothesized, the compound 4a has unsubstituted phenyl ring and exhibited poor growth anticancer inhibition properties with GI50 values of 23.76, 18.74, 12.86 and 21.63 µM in all cancer cell lines (Table 1). Substitution at the -para position of phenyl ring of 4a by halogen -bromo (4b) resulted moderate growth inhibition properties against MCF-7 cell line (Table 1, GI50 of 18.25 µM) while it showed potent growth inhibition against A549 and HepG-2 cell lines with GI50 values of.
2.97 and 3.62 µM respectively as compared to the standard doxorubicin (Table 1). Here, the activity was improved, this may be due to the -para bromo substituted phenyl ring. Introduction of another substituent by –chloro in –para position of phenyl ring of 4a brought the greatest improvement in the activity of the resulting compound 4b against MCF-7, A549 and HepG-2 cell lines (GI50 value of 2.63, 3.86 and 3.92 µM respectively) and indicating it was superior in potency against MCF-7 cell line compared to the doxorubicin (Table 1). This potency trend could be explained by the -para substituted halogen could have a potential role of drug–target binding affinity in above three cell lines.
In the next stage of biological activity, more compounds with substituted phenyl groups (electron-donating and electron-withdrawing) installed on the C4- and C5-positions of phenyl ring were prepared including substituents -OMe, -Me, -NO2 and -F (4d, 4e, 4f, 4 g, 4 h and 4i) diminished the potency against all four cell lines, (Table 1). Surprisingly, compound 4j has -methoxy substituent at -meta position of phenyl ring exhibited highest potent growth anticancer inhibition properties against MCF-7, A549 and HepG-2 cell lines (Table 1, GI50 of 1.82, 2.61 and 2.38 µM respectively) as compared to the doxorubicin (GI50 of 4.15, 3.26 and 3.87 µM respectively), this is mainly because of electrons donating effect of -methoxy group at –meta position could constructs π electrons interactions and contributing positively for the anti-proliferative activity (Table 1).
To learn the impact of the substituted-diathiazole compounds to potency, we synthesized compounds (8a-g) and screened for the anticancer activity. The substitution reaction on 4a by 5-amino-1,3,4-thiadiazole-2-thiol moiety resulted compound 8a in moderate reduction of antiproliferative activity as compared to 4a (Table 1). However, for –para substituted chloro compound 8b significantly increased anticancer inhibition properties against MCF-7 and HepG-2 cell lines (Table 1, GI50 value of 4.07 and 5.86 µM respectively) as compared to the compound 4b. This highlighted 5-amino-thiadiazole moiety and 4-substituted halogen of phenyl ring were playing a major role for the good activity by exploring binding sides to CDK9/Cyclin T1. In this series, compound 8d represented remarkable potential in MCF-7, A549 and HepG-2 cell lines (Table 1, GI50 of 2.98, 2.85, and 2.53 µM respectively) as compared to doxorubicin. Amazingly, compound 8f has strong electron-withdrawing substituent -para nitro of phenyl ring showed better activity as compared to 4f in MCF-7, A549 and HepG-2 cell lines with GI50 of 3.71, 4.64 and 3.46 µM respectively and still have comparable activity with doxorubicin (Table 1).
Thus, the compounds 8d and 8f have showed promising activity against MCF-7, A549 and HepG-2 cell lines as compared compounds (4a-i) and doxorubicin. This is indicating the crucial role of 5-amino-thiadiazole moiety in the potency.
Annexin V-FITC/PI assay
The early stages of apoptosis were monitored by Annexin V-FITC (apoptotic cell marker) and PI (necrotic cell marker) double staining. The staining method was used according to the Annexin FITC/PI staining kit (Invitrogen™, Thermo Fisher Scientific Inc.). Annexin V- FITC/PI assay was performed to inspect the extent of programmed cell death (Fig. 3, apoptosis) by the compounds 4c, 4j and 8f on breast carcinoma cell line (MCF-7). The cells were treated with increasing concentrations (5–10 μM) for 24 h and analyzed by flow cytometry.

Cell death (apoptosis) analysis of compounds 4c, 4j and 8f on breast carcinoma cell line (MCF-7). The cells were treated with increasing concentrations for 24 h and analyzed by flow cytometry. First quadrant Q1 represents necrotic cell, second quadrant Q2 represents late apoptotic, third quadrant Q3 represents live cells, and forth quadrant Q4 represents early apoptotic cells. The untreated cells have shown 88.26% of live cells, 0.21% death of cells due to necrosis, 0.18% due to the late apoptosis and 0.07% because of the early apoptosis. The experiments were repeated twice and representative data were presented
The untreated cells data has shown 88.26% of live cells, 0.21% death of cells due to necrosis, 0.18% due to the late apoptosis and 0.07% because of the early apoptosis (Fig. 3). The compound 4c has shown 62.43% and 42.19% of live cells at 5 and 10 μM concentration respectively. Whereas the necrosis is prominent cause of the cell growth inhibition at 5 μM and at 10 μM, the late apoptosis is the major reason for the cell death. Compound 4j has reduced the viable cell count to the 42.81% at 5 μM and at 10 μM it was 32.80% only. The cell death was majorly occurred due to the late apoptosis with 34.84% and 40.33% at the tested concentrations. Compound 4j has showed considerable reduction in the viable cell count as compared to 4c. This may be due to the electron-donating substituent (-methoxy) present on the phenyl ring at –meta position of 4j which have much better hydrophilic interactions due to availability of electrons. Whereas in compound 4c, the -chloro substituent present on phenyl ring at –para position leading minimum reduction of cell counts (Fig. 3). Interestingly, compound 8f has shown 59.08% and 46.14% of live cells at the tested concentrations. It has shown, the cell death of 16.99 and 14.62% at 5 μM due to necrosis and late apoptosis respectively and at 10 μM, the percentage of death was 12.44% and 37.15%. Thus, this underlined the role of substituted-diathiazole moiety in apoptosis.
Molecular modeling studies
The target prediction was done by using the Swiss Target Prediction and Pass Online tools. Based on the scores and ratio data from the two servers, potential anti-cancer targets like Cyclin-dependent kinase 9 (CDK9)/Cyclin T1–3BLQ signal transducer and activator of transcription 3 (STAT3) were selected [20, 21]. In addition, Sitemap tool of Glide was used for predicting the binding pockets and one of the predicted five was selected for docking studies based on the score and volume. Glide tool of the Maestro (Schrödinger Release 2023-2, New York, USA) was used for the exploring molecular docking studies. [22,23,24,25]
The docking studies of investigated compounds (4a-m) displayed XP GScores between − 7.777 and − 3.826 kcal/mol against CDK9/Cyclin T1 (1BLQ), providing the insights about their binding mode in the enzyme's binding cavity. Whereas verifying their potential affinity for the enzyme's binding cavity as compared to standard drug Flavopiridol as shown in Table 2 (Fig. 4). We have chosen reference drug Flavopiridol because it is an Orphan drug used for the treatment of acute myeloid leukemia as well as the treatment of arthritis. Flavopiridol is a known inhibitor of CDK9 and it was used as a standard inhibitor for comparing the docking results. STX-0119 is a co- crystalized ligand in STAT3 protein, it was used for validating the docking protocol and to compare the docking score and energy with the designed compounds. As per the data obtained from the experimentation, some of the designed molecules were found to possess better docking score with the studied protein molecules. Especially, compound 4 g was showed highest docking score – 7.777 kcal/mol against CDK9/Cyclin T1–1BLQ (Table 2) and found to interact with the binding site residues Cys106 (2.05, 2.66 Å) with H-bonding interactions and π-π stacking with Phe103 (Fig. 4a). As hypothesized, substitution of phenyl derivatives at C4 position of the thiazole ring afforded moderate to potent and relatively selective CDK9 inhibitors and other side of 5-amino-thiadiazole moiety could offers free -NH2 and -N groups as a key hydrogen bond donor and acceptor to interact with the backbone carbonyl and the -NH groups of the binding pockets respectively and possibly exhibited potential anticancer properties. Therefore, in this study we designed and synthesized six new 1,3,4-substituted-thiadiazole compounds (8b-g) and performed the docking studies for their potential anticancer properties. [12,13,14]

Binding modes of compounds 4g, 8b, 8d and 8g (from the top of the page figure a, b, c and d) with CDK9/Cyclin T1 and an important residues and interaction distances for key residues. CDK9/Cyclin T1 is shown in green color. The figure was prepared using Glide tool of the Maestro (Schrödinger, LLC, New York, USA)
In the first series of 4a to 4 m compounds, as discussed above compound 4 g has highest GScores (Table 2, − 7.777 kcal/mol), however experimental data revealed moderate growth in anticancer inhibition properties (Table 1). The compound 4c has also showed good GScores (Table 2, − 7.183 kcal/mol) and exhibited the strong binding interactions with the acidic residues of CDK9/Cyclin T1 and also the experimental data of anti-proliferative activity showed potent anticancer inhibition properties against MCF-7, A549 and HepG-2 cell lines (Table 1, 4c). Therefore, experimental data are in good agreement with docking data of compound 4c. Remarkably, the compound 4j showed highest potent growth anticancer inhibition properties experimentally as compared to all other compounds and the docking score for 4j against CDK9/Cyclin T1–1BLQ (Table 1, docking score = -6.075 kcal/mol and glide emodel = -51.949 kcal/mol) were comparable with the docking score of STX-0119 and CPB as shown in Table 2. This may be because of 4j has -methoxy substituent at –meta position of phenyl ring and it was moderately water soluble and has showed high GI absorption value with fulfilling Lipinski rule of five. However same isomeric compound 4d which has –methoxy substituent at –para position of phenyl ring and showed good dockings core (Table 2, − 7.464 kcal/mol) and found to be inactive practically (Tables 3 and 4). Similarly, for compounds 4e and 4f which showed acceptable docking score (Table 2, GScores − 6.376 and − 6.323 and kcal/mol) but experimental data indicating that they showed moderate anti-proliferative activity (Table 1, 4e and 4f). So in this series it was observed that -para substituted halogen and -meta substituted strong electron donating groups determined an improvement in the potency as compared to other compounds.
More interestingly, when compounds (4a-g) were extended by introducing amino-diathiazole moiety resulted highly potent compounds 8b, 8c, 8d, 8f and 8 g with highest docking score against CDK9/Cyclin T1–1BLQ inhibitor (Table 2, − 6.133, − 7.451, − 7.049, − 5.702 and − 7.324 kcal/mol respectively). However, the experimental data revealed that compound 8c showed moderate anticancer inhibition properties while compounds 8b, 8d and 8f showed potent anticancer inhibition properties (Table 1). The compound 8b has formed hydrophobic interaction with Lys and strong Hydrogen bonding with Leu and Glu amino acids residues (Fig. 4b). The compound 8d has -methoxy substituent at –para position of phenyl ring formed of strong Hydrogen bonding with Cys106 (2.82 Å and 2.29 Å) and Arg382(2.97 Å) as shown in Fig. 4c. Similarly, compound 8f has formed interactions with Glu107 (2.77 Å), Cys106 (2.24 Å), Phe103 (2.3 Å), Asp167 (2.19 Å) and Glu66 (2.51 Å) by strong H-bond and ionic interaction with Lys48 as shown in Fig. 4d. Additionally, all compound 8b, 8d and 8f have shown good pharmacokinetic properties as predicted by ADME (Tables 3 and 4).
In the drug likeness prediction and the pharmacokinetic property (ADME) calculations, it was found that all the compounds have molecular weight well below 500 Daltons and they may not face any difficulty to pass the biological membranes when given through oral route (Table 3). The same has been supported by the CLogP, where the compound have shown optimum partition coefficient, between 1.42 to 3.11. Whereas, all the compounds were having a minimum of 4 rotatable bonds required to undergo conformational changes during the binding to a protein target. The Hydrogen bond acceptors and donors were well within the accepted limits to meet the Lipinski rule of five. In addition to that the compounds were found to have high GI absorption and P-gp non-substrate nature (Table 4) makes them a viable choice to take them to next stage of exploration.
Thus, by experimental data, compounds 4j, 8b, 8d and 8f were found to be the potent anticancer agent. By the interaction energy, it was observed that these compounds have -bromo, -methoxy and -nitro substituent’s at -para position of phenyl ring of substituted-diathiazole compounds, which strongly assist the molecules to form potential bonds with the residues of the binding sites. It was proved that the docking calculations and the experimental data are close in proximity.
Conclusion
A series of novel 1,3,4-substituted-thiadiazole compounds (8b-g) designed and synthesized from 4-substituted-thiazol-2-chloroacetamides (4b-g) and fully characterized by spectrometric analysis methods (FT − IR, 1H NMR, 13C NMR, and mass). All synthesized compounds (4a-m) and (8a-f) exhibited selective anti-proliferative activity. Compounds 4j, 8d and 8f demonstrated promising anticancer activity against MCF-7, A549, HepG-2 and L02 all cancer cell lines. Especially compounds 4j and 8d exhibited the most potent anticancer inhibition properties with GI50 values of 1.82, 2.61, 2.38 μM and 2.98, 2.85, 2.53 μM against MCF-7, A549 and HepG-2 cell lines respectively as compared to Doxorubicin. Further molecular docking was revealed the biological activity. Thus, these compounds can provide promising starting points for further development of best anti-cancer agents.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and additionally its supplementary information file includes materials and methods, general experimental procedure, NMR data, NMR spectra, mass spectra and IR spectra].
References
GLOBOCAN (2020) New Global Cancer Data (UICC). https://www.uicc.org/news/globocan-2020-new-global-cancer-data. Accessed 27 Jul 2022.
Kalyanaraman B (2020) Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree. Redox Biolog 01394.
Mandal SR, Becker S, Klaus S. Targeting CDK9 for anti-cancer therapeutics. Cancers. 2021;13(9):2181.
Gonçalves M, Mignani S, Rodrigues J, Tomás H. A glance over doxorubicin based-nanotherapeutics: from proof-of-concept studies to solutions in the market. J Control Release. 2020;317:347–74.
Conroy A, Stockett DE, Walker D, Arkin MR, Hoch U, Fox JA, Hawtin RE. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother Pharmacol. 2009;64:723–32.
Albert TK, Rigault C, Eickhoff J, Baumgart K, Antrecht C, Klebl B, Mittler G, Meisterernst M. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br J Pharmacol. 2014;171:55–68.
Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Investig New Drugs. 2007;26:59–65.
Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD, Badros AZ, Popplewell L, Coutre S, Fox JA, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk 2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol. 2010;28:3015–22.
Dawood KM, Farghaly TA. Thiadiazole inhibitors: a patent review. Expert Opin Ther Pat. 2017;27:477–505.
Aliabadi A. 1,3,4-thiadiazole based anticancer agents. Anti-Cancer Agents Med Chem. 2016;16:1301–14.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Noblejas-López MdM, Gandullo-Sánchez L, Galán-Moya EM, López-Rosa R, Tébar-García D, Nieto-Jiménez C, Gómez-Juárez M, Burgos M, Pandiella A, Ocaña A. Antitumoral activity of a CDK9 PROTAC compound in HER2- positive breast cancer. Int J Mol Sci. 2022;23:5476.
Wakchaure SN, Shaikh SA et.al. (2024) Synthesis and Antimicrobial Evaluation of the 4-substituted-thiazol-2-chloroacetamidecompounds. Chemistry Select. Willey (article accepted and in press).
Yan G, Hao L, Niu Y, Huang W, Wang W, Xu F, Liang L, Wang C, Jin H, Xu P. 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamides as BACE1 inhibitors: synthesis, biological evaluation and docking studies. Eur J Med Chem. 2017;137:462–75.
Wolf L, Quoos N, Mayer JCP, de Souza D, Sauer AC, Meichtry L, Bortolotto V, Prigol M, Rodrigues OED, Dornelles L. Synthesis and free radical scavenging activity of 2-alkyl/arylchalcogenyl-N-(4-aryl-1,3-thiazol-2-yl)acetamides compounds. Tetrahedron Lett. 2016;57(9):1031–4.
Siddiqui N, Arshad MF, Khan SA. Synthesis of some new coumarin incorporated thiazolyl semicarbazones as anticonvulsants. Acta Poloniae Pharm Drug Res. 2009;66(2):161–7.
Özbek O, Gürdere MB. Synthesis and anticancer properties of 2- aminothiazole derivatives. Phosphorus Sulfur Silicon Related Elements. 2021;196(5):444–54.
Mishra D, Singh R, Rou C. A facile amidation of chloroacetyl chloride using DBU. Int J Chem Tech Res. 2017;10(3):365–72.
Balaji BS, Dala N. An expedient and rapid green chemical synthesis of N-chloroacetanilides and amides using acid chlorides under metal-free neutral conditions. Green Chem Lett Rev. 2018;11(4):552–8.
Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27(13):1907–18.
Becker S, Groner B, Müller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394(6689):145–51.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49.
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem. 2006;49(21):6177–96.
Pogodin PV, Lagunin AA, Filimonov DA, Poroikov VV. PASS Targets: Ligand-based multi-target computational system based on a public data and naïve Bayes approach. SAR QSAR Environ Res. 2015;26(10):783–93.
Bakchi B, Krishna AD, Sreecharan E, Ganesh VBJ, Niharika M, Maharshi S, Shaik AB. An overview on applications of Swiss ADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: a medicinal chemist’s perspective. J Mol Struct. 2022;1259: 132712.
Acknowledgements
We acknowledge the principal K.T.H.M. College and Kr. V. N. Naik Shikshan Prasarak Sanstha's Arts, Commerce and Science College, Department of Chemistry, Savitribai Phule Pune University (SPPU) for providing necessary laboratory facilities. We appreciate the provision of the required resources by the Director, SAIF, Chandigarh University for spectral analysis and NCL for providing sample for biological assay. We are expressing our sincere appreciation to SS Chobe of MGV College for their strong support and motivation in this research journey. SN Wakchaure, SA Shaikh, SR Labhade and RR Kale have provided the guidance for research and DD Bhanushali, KS Jain and HS Labhade were performed the organic synthesis experiments. Thanks to RR Alavala for assistance with the Laboratory facility for biological study. We are thankful to Mr. Sandeep Deshmukh, CMD of Delta Finochem Pvt. Ltd. for motivation, helpful discussion and providing LC-MS facility.
Funding
No funding has been received from any source.
Author information
Authors and Affiliations
Contributions
RR Alavala performed biological studies. SN Wakchaure, SA Shaikh, SS Chobe, SR Labhade, and RR Kale were constantly guided the projects. DD Bhanushali, KS Jain and HS Labhade were performed the organic synthesis experiments. All authors have gone through the final manuscript and approved it. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Additional figures and sections.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shaikh, S.A., Wakchaure, S.N., Labhade, S.R. et al. Synthesis, biological evaluation, and molecular docking of novel 1,3,4-substituted-thiadiazole derivatives as potential anticancer agent. BMC Chemistry 18, 119 (2024). https://doi.org/10.1186/s13065-024-01196-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-024-01196-1