
Abstract
Excessive cell proliferation due to cell cycle disorders is one of the hallmarks of breast cancer. Cyclin-dependent kinases (CDKs), which are involved in the transition of the cell cycle from G1 phase to S phase by combining CDKs with cyclin, are considered promising targets with broad therapeutic potential based on their critical role in cell cycle regulation. Pharmacological evidence has shown that abnormal cell cycle due to the overexpression of CDK6 is responsible for the hyperproliferation of cancer cells. Blocking CDK6 expression inhibits tumour survival and growth. Therefore, CDK6 can be regarded as a potential target for anticancer therapeutics. Thus, small molecules that can be considered CDK inhibitors have been developed into promising anticancer drugs. In this study, combined structure-based and ligand-based in silicon models were created to identify new chemical entities against CDK6 with the appropriate pharmacokinetic properties. The database used to screen drug-like compounds in this thesis was based on the best E-pharmacophore hypothesis and the best ligand-based drug hypothesis. As a result, 147 common compounds were identified by further molecular docking. Surprisingly, the in vitro evaluation results of 20 of those compounds showed that the two had good CDK6 inhibitory effects. The best compound was subjected to kinase panel screening, followed by molecular dynamic simulations. The 50-ns MD studies revealed the pivotal role of VAL101 in the binding of inhibitors to CDK6. Overall, the identification of two new chemical entities with CDK6 inhibitory activity demonstrated the feasibility and potential of the new method.
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Breast cancer, which accounts for 1/4 of all malignant tumours in the world [1], is a common and high-risk malignant tumour for women, and breast cancer is an active research area that has attracted substantial attention from scholars. The current literature on breast cancer covers all aspects of prevention and early detection as well as treatment and beyond. Studies have shown that the abnormal cell cycle caused by the abnormal effects of proto-oncogenes and tumour suppressor genes directly manifests as infinite growth of breast cancer cells, which is the most common and basic biological characteristic of breast cancer. Therefore, controlling abnormal cell cycle is the most direct way to effectively inhibit tumour growth.
Cell cycle regulation is a complex biological process involving many genes and proteins, which together form an extensive network of signalling molecules. CDKs belong to the serine/threonine protein kinase family, and these proteins are at the heart of cell cycle regulation [2]. CDKs have various isomers and are known for their broad-spectrum therapeutic potential against uncontrolled regulation. The CDK4/6-CycD-INk4-pRb-E2F signalling pathway plays an important role in promoting the transition of the cell cycle from the G1 phase to the S phase. The amplification of the CDK4/6 gene caused by mutation or cell cycle inhibitor deficiency can be observed in multiple tumours. It was reported that CDK6 has a unique function different from that of CDK4, and it is involved in cell metabolism [3,4,5] and cell differentiation [6, 7], transcription, and regulation of DNA repair [6, 7]. CDK6 inhibitors could limit the survival and growth of tumour cells, promote the apoptosis of tumour cells and improve the drug sensitivity of tumour cells. Excitingly, it was demonstrated that FDA-approved CDK6 inhibitors have a good therapeutic effect on breast cancer. In general, CDK6 is a promising target for drug development, and efficient drug design is very meaningful for the treatment of breast cancer in the future.
CDK6 has attracted much attention for many decades, and many inhibitors have been developed. To date, three approved drugs for breast cancer have shown significant clinical activities [8]. Moreover, palbociclib and ribociclib, which target pancreatic cancer and colorectal cancer, also represent a promising clinical therapeutic strategy. In addition, some CDK6 inhibitors, such as 7-hydroxystaurosporine, FLX-925, lerociclib, and alvocidib hydrochloride, are currently in clinical trials. Furthermore, some CDK6 inhibitors are pyrimidine derivatives, such as pyrimidine, pyridine, pyrimidinethiophene, aminopyrimidine, bisaminopyrimidine, and pyrimidineindole derivatives [9,10,11,12]. Other CDK6 inhibitors have structures such as fascaplysin, dioxothiazol, and lycoline or their derivatives [13,14,15].
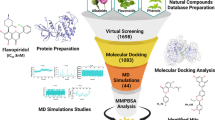
In the present study, as shown in Fig. 1, the X-ray crystal structures of the target proteins in complex with CDK6 inhibitors and various CDK6 inhibitors were applied to design structure-based and ligand-based models. Ligand-based pharmacophores were built based on known CDK6 inhibitors with common pharmacophore features. The E-pharmacophore model was built on the receptor–ligand complex with Glide XP scoring terms. E-pharmacophore is a hypothesis based on the complementarity of receptor and ligand features. These models generated in silicon were applied in parallel to screen the drug-like databases ChemDiv and ChemBridge. Consequently, molecular docking was performed on the molecules identified by pharmacophore screening. Considering the key residues, good ADMET properties, binding energy, and structural diversity, several compounds were selected for biological activity tests. To study the selectivity of the hit compound, the best candidate with high activity was subjected to kinase panel screening. Ultimately, MD simulations were carried out to determine the accurate binding mode between the protein and the ligand. Overall, the results showed the success of our virtual screening method to identify new CDK6 inhibitors, and this scaffold is worthy of further optimization studies.

Virtual screening workflow
Materials and methods
Protein preparation and binding site analysis
The RCSB Protein Database (PDB) provided the X-ray structures of the following seven Homo sapien CDK6 proteins and their respective inhibitors, namely, 3NUP, 3NUX, 4AUA, 4EZ5, 5L2I, 5L2S, and 5L2T. As shown in Table 1, these small-molecule inhibitors derived from high-resolution complexes in PDB have inhibitory activities ranging from 10 to 7200 nM. All the selected proteins were then prepared using Protein Preparation Wizard (Maestro module of Schrödinger) with the default parameters, including the elimination of water molecules, the addition of hydrogen atoms, and filling in missing residues. Restraint minimization was carried out with the OPLS3 force field with a root-mean-squared deviation (RMSD) convergence to 0.3 Å [16]. Among these candidate proteins, the optimal protein was selected based on the results of Glide Extra precision (XP) Docking results and SiteMap simulation. These native ligands were prepared and re-docked to the ATP-binding site of CDK6 with Glide XP docking, and the RMSD between the conformations of bound and docked inhibitors was calculated. A smaller RMSD between the conformations of the docked and bound compounds indicated a greater likelihood of retrieving bioactive conformations of the compounds. SiteMap is a tool for recognizing, visualizing, and evaluating protein binding sites in Schrödinger. SiteMap provided an algorithm for binding site identification, and the evaluation can help researchers locate binding sites with a high degree of confidence and predict the druggability of those sites [17]. SiteScore was used to assess a site’s propensity for ligand binding, and the docking site was considered druggable only when the SiteScore was > 1.0 [18].
E-pharmacophore generation and validation
In addition to providing ligand information, E-pharmacophores with XP description can be used to further determine the features that contribute the most to the binding energy. The Pharmacophore Alignment and Scoring Engine (PHASE) of the Schrödinger module was employed for hypothesis generation with six default chemical features, including a positive ionizable group (P), a negative ionizable group (N), an aromatic ring (R), a hydrogen bond acceptor (A), a hydrogen bond donor (D), and a hydrophobic region (H) [19]. For E-pharmacophore generation, all the refined cocrystal ligands were re-docked onto the corresponding prepared protein structures using XP docking with standard van der Waals scaling at 0.8 and a partial charge cut-off of 0.15. Initially, seven pharmacophore models were designed for all the crystal structures. Then, the accuracy of these E-pharmacophore hypotheses was validated with a dataset consisting of active and inactive compounds.
A dataset comprising of 88 active compounds and 1000 inactive compounds was used to validate these generated pharmacophore hypotheses. These 88 active compounds are established CDK6 inhibitors selected from the ChemBL database. The inactive compounds (decoys) were retrieved using Decoyfinder2.0 software. We focused on robust initial enhancement (RIE), Boltzmann-enhanced discrimination of receiver operating characteristic (BEDROC), receiver operating characteristic (ROC), and area under the accumulation curve (AUCU) metrics to assess the quality of the pharmacophores generated in this work. RIE was selected as the first metric, which is less susceptible to the length of the dataset than enrichment factor (EF). It represented active ranks by weighting a continuously decreasing exponential term [20]. The second metric, BEDROC, generated from the ROC and possessing a probabilistic meaning [20], also used to evaluate the significance of results. In addition, AUCU and ROC are well-recognized standards to quantify the reliability of pharmacophore models, and they were used to estimate the ability of the hypothesis to identify active and inactive compounds [20]. Based on the criteria above, the E-pharmacophore hypotheses without discriminating ability were discarded, and the top-ranked pharmacophore models were used for screening the filtered databases.
Ligand-based pharmacophore generation
Although structure-based computational studies are satisfactory, ligand-based studies are also important. Ligand-based 3D pharmacophores, the spatial arrangement of chemical features common to at least two active ligands, are used to propose the essential interactions for ligand binding and were used to identify potential candidates from these databases. In this study, a ligand-based 3D pharmacophore model was built with PHASE in Schrödinger using another 92 inhibitors collected from the literature [14, 21, 22] and ChEMBL database (https://www.ebi.ac.uk/chembl/) with IC50 values ranging from 0.01 to 30,000 nM. Ligand-based pharmacophore generation included ligand preparation, pharmacophore site creation, common pharmacophore identification, hypothesis scoring, and QSAR model analysis.
Before pharmacophore building, the structures of these inhibitors were sketched and prepared using the default setting of LigPrep, and the energy of the inhibitors was minimized with the OPLS3 force field. Conformers were explored through macromodel combined with the MCMM/LMOD (mixed torsional/low-mode) method, which is the most powerful conformation searching method currently available. The maximum number of conformers was 1000 per structure. The conformers were filtered through a relative energy threshold of 21 kJ/mol and deviation beyond 0.5. Subsequently, these conformers were minimized with the Polak–Ribiere Conjugate Gradient (PRCG). Then, the IC50 values of these compounds were converted into pIC50 values. Compounds with pIC50 value greater than 6.8 are considered active, compounds with a pIC50 value less than 6.0 were considered inactive, and the rest were moderately active compounds. Therefore, these compounds were separated into active, inactive, and moderately active compounds based on these threshold values. Thereafter, these prepared ligands were subjected to common pharmacophore hypothesis construction, and the generated hypotheses were ranked by several score functions.
The generated ligand-based pharmacophores that survived the scoring process were subsequently subjected to 3D-QSAR analysis in PHASE to validate the developed pharmacophores. First, with these aligned ligands randomly split into training and test sets at a ratio of 3:1, atom-based QSAR analysis was performed with five PLS factors and the built parameters [23, 24]. Then, the training set compounds were considered for 3D-QSAR model generation with a 1.00 Å grid spacing, and the test compounds were used to validate the QSAR models. Variables with |t value| < 2.0 were eliminated for good predictions of the test set compounds, and 36 ligands were eliminated for leave-more-out (LMO) cross-validation. LMO-CV, where more than one chemical was left out of the validation at a time, had a stronger CV than LOO-CV [25]. The correlation between the chemical structural components (independent variable-dependent components) was also analysed for these developed ligand-based pharmacophores. The best hypothesis was further validated and selected based on the values of the correlation coefficients of the training set (R2) and test set (Q2).
Pharmacophore-based virtual screening
These selected pharmacophores were applied to screen the library of compounds. The compound libraries used for virtual screening were prepared using the LigPrep module of Schrödinger with the OPLS3 force field. During this process, low-energy 3D structures were generated. The two prepared compound libraries were then filtered using Lipinski’s rule of five to retain the drug-like molecules. [26]. The retrieved hits remained when their fitness value was better than 1.8 because the fitness values of the approved CDK6 drugs are better than 1.7 (palbociclib: 1.86, abemaciclib: 1.780, and ribociclib: 1.738). All the retrieved hits from E-pharmacophores and 3D-pharmacophore were further subjected to molecular docking refinement.
Docking-based virtual screening
These retrieved inhibitors were prepared and docked in the active site of CDK6 using the HTVS-SP -XP virtual screening workflow of Glide. High-throughput virtual screening (HTVS) docking is intended for the rapid screening of very large numbers of ligands. HTVS has a much more restricted conformation sampling than SP docking and cannot be used with score-in-place and predetermined values. Glide SP is a protocol for screening ligands of unknown quality, while Glide XP helps determine all reasonable conformations of low-energy conformers at the designated binding site and is considered a refinement tool for eliminating false-positive findings. Extraprecision (XP) docking and scoring is a more powerful and discriminating procedure that takes longer to run than SP. XP is designed to be used on ligand poses that have been determined to be high-scoring using SP docking. Therefore, HTVS, SP and XP docking were carried out for molecular docking screening, and compounds with docking scores of better than − 9.0 were retained (the average docking score of known CDK6 inhibitors was − 9.0). Considering the minimum docking score, structural diversity, and interactions with the key residue, 35 compounds were subjected to IFD docking, ADMET property prediction and post-docking Prime MM/GBSA evaluation.
Induced-fit docking
Induced-fit docking is a mixed molecular docking and dynamics method in which the receptor is flexible and the ligand is rigid during the docking study [27, 28]. It aims to improve the docking of ligands and find the optimal binding model of proteins and small molecules. First, the 5L2S protein was subjected to energy minimization using the OPLS3 force field with an implicit solvation model. Then, a grid box was generated at the centroid of the ligand, which was similar in size to the workspace ligand. After that, these hits were docked to the rigid protein with a van der Waals (VDW) radius scaling of 0.5 for the atoms in both the protein and the ligand. Residues within 5.0 Å of the corresponding ligand positions were included in the Prime refinement. A final round of Glide XP docking and scoring was carried out for the shortlisted protein structures. The IFD score indicated the binding energy for the protein–ligand complex, and a higher negative IFD score means more favourable binding with the target.
Free binding energy calculation
The binding free energies of the selected docking complexes were calculated by using the generalized Born surface area of molecular mechanics with the Prime module of Schrödinger. Prime MM-GBSA is a well-known method to determine solvation free energy resulting from electrostatic effects within a generalized Born model. It has been used for the MD simulations, energy minimization, protein–ligand binding affinity predictions, and identification of the important residues for the protein–protein interactions. [29]. In this study, Prime MM/GBSA with VSGB 2.0 models and OPLS3 force field was used for ligand binding and ligand strain energy estimation. The structures of the complexes from the IFD docking with good docking scores were further considered for binding free energy calculations, which were calculated as follows.
where ΔE, ΔG, and ΔGSA represent the minimized energy, solvation free energy, and surface area energy of the complex, protein, and ligand, respectively.
ADMET prediction
Effective and safe drugs exhibit high potency, affinity, and selectivity against the molecular target, along with adequate absorption, distribution, metabolism, excretion, and tolerable toxicity (ADMET). Evaluation of the ADMET properties is considered indispensable because it can significantly improve the success rate of drug development and reduce development costs in drug discovery. The QikProp model in Schrödinger was used to evaluate the pharmacological properties of the candidate compounds to exclude compounds with unsuitable ADMET properties. The QikProp application predicts a number of ADMET parameters and identifies drug-like compounds on the basis of values obtained for 95% of known drugs. The MW (molecular weight), QPlogS (predicted aqueous solubility), and RO5 (Lipinski’s rule of five) were used for drug-likeness evaluation. The gut-blood barrier (QPPCaco) and blood–brain barrier (QPPMDCK), skin permeability, and predicted IC50 value for the blockage of HERG K+ channels were also predicted.
Cell proliferation inhibition
MCF-7 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in Gibco™ Dulbecco’s modified Eagle’s medium under aseptic conditions in a 37 °C humidified CO2 incubator supplemented in 10% heat-inactivated foetal bovine serum (Biological Industries). Following two generations, the cells were treated with 25% trypsin–EDTA solution and then seeded in 96-well plates with 8 × 103 cells per well in 100 μL of complete culture medium for an MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) assay.
The antitumour effects of the studied compounds were tested on MCF-7 cell lines with an MTT assay as previously described. Multiple studies have indicated that the overexpression of CDK6 in cancer cells and their oncogenesis could be disrupted by CDK6 inhibitors. In short, after 24 h of incubation to reattach and recover, the seeded cells were treated with the test compounds at an initial concentration of 30 µM in triplicate. The two test compounds were first dissolved in high-grade DMSO and then diluted in culture medium to five different concentrations (1.875 μM, 3.75 μM, 7.5 μM, 15 μM, and 30 μM).
To avoid bystander cytotoxicity, the final DMSO concentration was kept at less than 0.1%. Three wells were left untreated as cell-based negative controls, and three wells of cell culture medium were left as blanks. Palbociclib at different concentrations was used as the positive control. After 72 h of incubation, the media were removed, and then, the cells, including the controls, were treated with DMEM containing MTT (0.5 mg/mL) solution, incubated for 3 h at room temperature, and inspected periodically for purple formazan precipitate. With the media discarded, 100 μL of DMSO was added to dissolve the formazan crystals. The OD value (optical density) of each well was measured at 490 nm with a microplate reader following 30 min of incubation at 37 °C in the dark. Finally, their IC50 values were determined by dose–response curves, and data analysis was performed with the GraphPad Prism package.
CDK6 kinase assay
The CDK6 kinase assay was performed by Shanghai Wellfeng Biotech. In this assay, the CDK6/cyclin D3 activity was determined with ELISA with the enzyme, substrate, ATP and inhibitors diluted in kinase buffer for both the reaction mixture (kinase reaction in the presence of substrate) and blank control (kinase reaction in the absence of substrate). The reaction was initiated by adding 3 μL of ATP into 30 μL of buffer solution containing HIT8 and HIT14 at different concentrations or DMSO (1 μL), CDK6/cyclin D3 (0.20 nM), HEPES-Na (50 mM, pH 7.5), MgCl2 (5 mM), biotin-pRb (773-924, 200 nM), BSA (0.05%), Tween-20 (0.02%), and DTT (1 mM). Then, the reaction was quenched with EDTA-Na (120 mM, pH 8.0, 10 μL) and incubated for 2 h. Following incubation of the detection solution (40 μL) containing Eu-W1024 anti-rabbit IgG antibody (2 nM), antiphospho-pRb (S780) antibody (143 ng/mL), and SA-APC (40 nM) in detection buffer for 30 min, the plate luminescence was recorded with PE EnSpire. The IC50 values were determined using GraphPad Prism Software.
Kinase panel screening
The best compound was subjected to kinase panel screening by Eurofins Pharma Discovery Services. The optimal compound, HIT14, was tested for its ability to inhibit 105 kinases at a concentration of 2 μM. First, the reaction buffer, which contained 0.02% Brij35, 0.1 mM Na3VO4, 10 mM MgCl2, 2 mM DTT, 1 mM EGTA, 0.02 mg/mL BSA, 1% DMSO, and 20 mM HEPES (pH 7.5), was prepared. Afterwards, the required cofactors were added to each kinase reaction separately as needed. In the enzyme reaction procedure, the required cofactors were added to fresh buffer solution, and then the selected kinases at a concentration of 20 μM were added. After gently mixing all the components, the test compound (HIT14) was dissolved in DMSO and added to the reaction mixture. 105-ATP (specific activity 500 μCi/μL) was added to initiate the reaction, and the mixture was incubated at room temperature for 2 h. In the initial screening of over 105 kinases, HIT14 was tested by a single-dose duplicate prepared at a concentration of 2 μM. Staurosporine was used as a control compound in a 5-dose IC50 mode with 10-fold serial dilutions starting at 20 μM. The reaction was carried out with ATP at a concentration of 10 μM.
Molecular dynamic simulation
To identify accurate combination modes and the key amino acid residues for CDK6 inhibition, MD simulation studies were applied to the docked pose of the hits. MD simulations provide details regarding the motion of individual atoms in a molecule [30, 31]. MD simulations help sample the configurational space with atomic force fields within nanoseconds, thus revealing molecular conformations and facilitating the evaluation of their interactions with water, ions or low-molecular-weight ligands. [32]. In the present study, the most promising candidate compounds were subjected to MD simulations. The solvation system including the solute and solvent water molecules necessary to neutralize the system was generated via the System Builder. The system was an explicit solvent with the TIP3P model in a cubic box. There were 10 Å buffer regions between intramolecular protein atoms and the box sides to specify the conformation and the size of the repeating unit. Moreover, 2 Na+ counter ions were added to neutralize the system, and 0.15 M NaCl solution was used to mimic the physiological environment. The complex was restricted with a force constant of 1 kcal/mol Å2, and minimization was carried out using the limited memory Broyden–Fletcher–Goldfarb–Shanno algorithm with the initial steepest descent processed until the gradient threshold was 25 kcal/mol Å2. Similarly, the protein was restricted only at 0.1 kcal/mol Å2, and minimization was repeated. Furthermore, the system was gradually heated from 0 to 300 K, with a force constant of 2.0 kcal/mol Å2 applied to the complexes throughout the heating process. Under the constant temperature/constant pressure ensemble, the limiting force constant was gradually reduced, and the composites were sequentially simulated with force constants of 2.0, 1.5, 1.0, 0.5, and 0.1 kcal/mol Å2 to simulate 500 ps. Since then, a 50-ns unrestricted molecular dynamics simulation was carried out with the OPLS3 force field in Schrödinger Desmond. Berendsen and Martina–Tobias–Klein barostats were used to maintain the isothermal-isobaric ensemble at 300 K and 1 atm. Moreover, short-term bonds or non-bonding interactions could be obtained using the RESPA integrator with a time step at 2.0 fs. A long-range interaction cut-off radius of 9 Å was used in combination with the smooth particle mesh Ewald summation. Finally, the structural and configurational trajectories were recorded every 5 ps by visually inspecting the 3D structures.
Results and discussion
Protein preparation and binding site analysis
Seven cocrystal structures of different resolutions were retrieved from the PDB. The ligands contained diverse structural scaffolds. Protein preparation wizard was used to ensure the structural correctness of the proteins in the OPLS3 force field. The refined protein structures obtained are shown in Fig. S1. Through the Glide extra precise (XP) docking model in Schrödinger, all the refined native ligands were docked at the respective prepared protein structures. As shown in Table 2, the protein with PDB-ID code 5L2S was selected for further docking studies since the 5L2S protein showed the highest identification ability with an average RMSD of 0.86 Å and a better SiteScore (1.108) and XP docking score for its native ligand (-12 kcal/mol) (Table S1).
E-pharmacophore
The E-pharmacophore approach, integrating the pharmacophore concept with protein–ligand XP energetic descriptions, was explored to generate the E-pharmacophore hypothesis. In this thesis, all the crystal structures of CDK6 were applied to develop the pharmacophore models. At least four out of seven pharmacophore sites were selected for matching. Information about these energetically beneficial sites, including certain specific interactions, is potentially beneficial for the design of new inhibitors. The E-pharmacophore method was validated in terms of the RIE, ROC, and BEDROC parameters (alpha = 160.9, alpha*Ra = 13.0140; alpha = 8.0, alpha*Ra = 0.6471; alpha = 20.0, alpha*Ra = 1.6176, respectively) based on the retrieval rate of the active ligands in the database containing inactive compounds. Overall, the enrichment analysis results indicated that the pharmacophore model is applicable for subsequent docking screening. As shown in Table 3 and Fig. 2, the best pharmacophore, called ‘hypothesis 3NUX’, presented good screening performance with the highest RIE value (8.58), BEDROC value (above 0.84), and optical EF1% value (11.24). Moreover, in summary, this model can distinguish the active and inactive compounds well based on the receiver operating characteristic (ROC) graphs (0.83) and area under the ROC curve (0.87). Therefore, hypothesis 3NUX (HDDRR; Fig. 2) was selected for subsequent database screening.

E-pharmacophore properties. a E-pharmacophore feature of 3NUX, H7 represented hydrophobic site; R10, R11 represented aromatic ring; D4, D5 represented H-bond donor. b Distance of E-pharmacophore features, c ROC curve of pharmacophore hypothesis 3NUX, d EF1% of hypothesis
Ligand-based pharmacophore
In view of the built-in pharmacophore features, a number of pharmacophore hypotheses with different features were developed and ranked by survival score, which took into account the vector, volume, survival inactive score, selectivity, and the number of matches [33]. Atom-based 3D-QSAR studies were performed to validate and select the developed pharmacophore. As mentioned above, the initial data were randomly separated into training and testing sets. Seventy-two compounds were in the training set used to develop the 3D-QSAR model, and the remaining 24 molecules were used as the test set to verify the generated model. In addition, the ‘leave more out’ (LMO) cross-validation method was applied to evaluate the predictive ability of the 3D-QSAR models. As a result, the best hypothesis code AADHRR.81 (Fig. 3a) was selected based on its good prediction of both the training and test sets with a high R2 value of 0.9362, a high Q2 value of 0.8229, and a low SD value of 0.2984 (in Table 4). The distances between the pharmacophore features in the hypothesis are also shown in Fig. 3a, and the plots of the actual vs predicted activities of the compounds in the dataset are depicted in Fig. 3b. As a consequence, hypothesis AADHRR.81 was selected for novel compound screening from the ChemDIV and ChemBridge databases.

a Measurement of AADHRRR.81 pharmacophoric site, pharmacophore features were represented by light red sphere for hydrogen bond acceptor (A) with the arrows pointing in the direction of lone pairs, green sphere for hydrophobic regions (H), and orange torus for aromatic rings (R), b Scatter plot of the predicted activity (pIC50) versus activity (experiment pIC50) on PLS factor
Pharmacophore-based virtual screening
The E-pharmacophore model and ligand-based pharmacophore were combined to screen the databases. With distance matching tolerances of 2.0 Å, the identified molecules were ranked on their fitness score (range from 0 to 3) with a cut-off > 1.7. The fitness score fully considered the alignment score, volume score, vector score, and RMSD of the aligned ligand. In the light of the two pharmacophores described above, 108,736 and 37,201 compounds against CDK6 were screened from the compound libraries in parallel (Table 5).
Docking-based virtual screening
The molecules with a high fitness score were deemed active inhibitors. Separated docking screening approaches were applied to reduce the false-positive rate. The retrieved compounds were further docked on the crystal structure of CDK6 (5L2S) using the built-in scoring function, and compounds with a docking score higher than -9.0 were filtered from the virtual screening workflow. The number of hits retrieved from pharmacophore and molecular docking is presented in Table 5. Next, by further analysing the interactions between these compounds and key residues, 147 common compounds were identified and selected. Notably, these compounds interacted with the key VAL101 residue in the ATP-binding pocket [21, 22, 34]. The hits retrieved in this virtual screening workflow were subsequently compared with the literature using the SciFinder database. Overall, 35 compounds that have not been reported to have with antitumour or kinase inhibitory effects were selected through this method.
IFD docking
Induced-fit docking (IFD) simulations were carried out using the novel 35 candidate molecules obtained from the docking simulations. The binding of each molecule to the protein was judged based on the IFD score, which was similar to the Glide XP score but with some differences. Although their conformations significantly different from those produced by rigid docking, the IFD scores, ranging from − 448.43 to − 524.29 kcal/mol (in Table S3), indicating a good binding model.
ADMET analysis
Analysis of the ADMET properties improves the quality of drug development by reducing costs, thereby increasing the success rate. In the present study, the ADMET properties of each candidate compound were analysed. As shown in Table S5, QP logPo/w and QPlogS were viewed as the first standards, representing the absorption and distribution of the drug within the body, respectively. Furthermore, QPPCaco, QPlogHERG, QPlogBB, and QPPMDCK are critical parameters. We explored whether the compounds violated the RO5 parameter, and their advantageous pharmacogenetic properties revealed their potential as candidate hits. For instance, 3 hits (numbered C2, C3 and C23) violated one of Lipinski’s rules of five. In addition, some compounds, such as C23 and C30, showed undesirable water solubility. The skin permeability of some compounds, such as C2, C3, C21, C23, and C28, needed to be improved. Consequently, 20 compounds with good pharmacokinetics and appropriate toxicological properties were identified.
Free binding energy
Post-docking Prime MM-GBSA was carried out to estimate the complex-binding free energies (ΔGbind values) and elucidate the binding affinities. This method considered the effects of desolvation and thermodynamics [35]. The ΔGbind values varied slightly from those determined through docking, as various types of energy were considered. A Prime energy calculation produces the Prime Energy and in addition generates individual contributions to the energy of various types, such as coulomb energy, covalent binding energy, Van der Waals energy, and generalized Born electrostatic solvation energy. The coulomb interactions fluctuated because a mixture of charged and neutral residues was considered. The Vdw and Lipo values essentially accounted for VDW and hydrophobic interactions during inhibition. The molecules retrieved from the docking screening displayed good binding free energies, and some of these hits displayed ΔGbind values higher than that of the native ligand of 5L2S (− 59.409 kcal/mol) (Table S5). The hydrogen bond energy indicated the contributions of hydrogen bonds, which were significant in the interactions of some compounds, such as HIT3, HIT10, and HIT18, with the target protein. In general, the binding free energy calculation based on the Prime MM-GBSA method also supports the stability of the HIT-CDK6 complexes.
Anti-tumour and kinase assay
The 20 compounds retrieved in this study were purchased, and a tumour suppression assay was carried out with a primary concentration of 30 µM. As shown in Table 6 and Fig. 4a, two compounds out of these showed cell inhibition relative to the control group in the concentration range of 1.875–30 μM, and their IC50 values were 21.13 µM and 12.32 µM. Then, these two compounds were tested for their CDK6 kinase inhibitory potential at concentrations of 10 µM, 5 µM, 2.5 µM, 1.25 µM, and 0.625 µM. A dose–response curve with increasing doses is shown in Fig. 4b. As a result, HIT8 (IC50 = 3.22 µM) and HIT14 (IC50 = 1.48 µM) could be promising hits for optimization against the CDK6 enzyme.

Inhibition effects of HIT8 and HIT14 a cell inhibition of HIT8 and HIT14, b graph plot of CDK6 inhibition
Thus, two inhibitors targeting CDK6 were obtained, and their binding modes are shown in Fig. 5. All of the docking results suggested that these compounds, which are structurally distinct, were located in the ATP-binding pocket, and they may show specific ATP competitive binding. Similar to previous reports, each compound formed one or more hydrogen bonds with the VAL101 residue [21, 22, 34]. As plotted in Fig. 5a, b, the novel compound HIT8 formed a H-bond with the main chain NH and the backbone carbonyl of VAL101, and the distal benzene of HIT8 reached a more polar and solvent-exposed region in the ATP-binding pocket, which consisted of amino acid residues such as ASP104 and THR107. Compared with the binding modes of the crystallized ligand of 5L2S (the structure is shown in Table 1), both the key hydrogens in the hinge region and the key interactions with the core of the scaffold were maintained. As shown in 5C and 5D, there were two hydrogen bonds with the key amino acid VAL101. In addition, the distal benzene of HIT14 can reach a more polar and solvent-exposed region and form a polar interaction with ASP104 and THR107. Their binding model also suggested that the polar group-substituted benzene ring may enhance the strength of the H-bond with the amino acids LYS43 and ASP163, which might stabilize the protein–ligand binding model. The above molecular docking analyses revealed that the identified hits had excellent binding modes with the target protein and would provide meaningful lead compounds for novel CDK6 inhibitor discovery.

3D and 2D map of the binding pose of the best 2 compounds from IFD docking results
Kinase panel screening
To investigate the possible kinase inhibitory activity of the new compound, HIT14 was tested against a panel of 105 different kinases at Eurofins Pharma Discovery Services. The screening results revealed remarkable inhibitory activity against CDK6 kinase. The compound was initially tested at a single-dose concentration of 2 μM. At this concentration, 78% inhibition of the enzymatic activity of CDK6 kinase was observed. Since CDK4 and CDK6 have extensive homology, this novel compound also inhibited CDK4 (Fig. 6, Table 7).

Summary of kinase inhibitory profile of HIT14 at 2 μM over a panel of 105 kinases
Molecular dynamic simulation
The binding pocket primarily comprised ILE19, GLU21, TYR24, VAL27, ALA41, LYS43, PHE98, GLU99, HIS100, VAL101, ASP102, GLN103, ASP104, ASP145, LYS147, GLN149, ASN150, LEU152, and ALA162. The best compound, HIT14, the binding mode of which is shown in Fig. 5c, d, was selected for the MD study. Compared with molecular docking, MD simulations provide a more accurate estimation of binding affinities and binding poses, as it is focused on the dynamic aspects of different protein conformational stages, namely, snapshots. It was observed that the novel compound formed stable interactions with the CDK6 protein in the 50 ns MD simulations. As shown in the RMSD plot (Fig. 7A), although fluctuations occurred, which may be caused by intramolecular H-bond interactions (seen in Figure S3), the protein was stable approximately 2 ns after the simulation, and the system finally reached equilibrium. Multiple H-bonds between the ligand and VAL101 were detected in the MD simulations (Fig. 7b, c). In addition, hydrogen bonds between the CDK6 protein and ILE19 and GLN149 mediated by a water molecule were detected. Other amino acid residues showed hydrophobic interactions with the ligands. In addition to hydrophobic contacts, there existed additional hydrogen bonds to HIS100 and a water bridge to ILE19 (Fig. 7b). Additionally, H-bonds with VAL101 were present for over 50% of the MD simulation time, and the water bridge with GLN149 and π-π stacking with PHE98 occurred in 11% and 24% of the total simulation time, respectively (Fig. 7c). From the above analysis, it could be concluded that compounds stably bound in the ATP-binding pocket of the CDK6 protein, and VAL101 was significant for protein–ligand stabilization.

Protein ligand interaction in MD simulation a RMSD of complex 14, b stacked bar charts of protein–ligand 14, c ligand atom of hit14 interactions with the protein residues
Conclusion
In summary, two novel CDK6 inhibitors with diverse structures have been identified from the compound libraries ChemDiv and ChemBridge, which contain 50,000 drug-like compounds, through an approach combining structure-based and ligand-based computational studies. The computational methods were applied in parallel to fetch a maximum number of compounds from each independent methodology. The availability of X-ray crystal structures and a number of known inhibitors of CDK6 provide a gateway for performing efficient in silico studies models on this target.
In this study, energetically optimized pharmacophore models were generated on the cocrystal structures of all proteins and validated via datasets containing active and inactive compounds. As a consequence, the pharmacophore hypothesis derived from the crystal structure coded 3NUX (HDDRR) displayed good identification ability with an RIE value of 8.58 and BEDROC values of 0.90, 0.865, and 0.841. Furthermore, the model could distinguish active compounds from inactive compounds because the ROC and AUCU were up to 0.8. In addition, ligand-based pharmacophore code AADHRRR.81, which had the highest survival score (3.727), R2 value (0.9362), and Q2 value (0.8229) and a low SD value (0.2984), was generated and selected. After that, to retrieve the maximum number of compounds, these hypothesis models were used to screen the libraries in parallel. Then, a multilayer molecular docking screening workflow was performed to screen the compounds retrieved from the pharmacophore model. To obtain the maximum sensitivity (true positive rate) of the model, the compounds screened from the E-pharmacophore and 3D-QSAR pharmacophore models were separately subjected to molecular docking during the virtual screening process. In this process, a total of 20 new compounds with the characteristics of good pharmacophore features, ADMET properties, reliable binding modes and scaffold diversity were screened. These compounds were selected for the in vitro antitumour assay and CDK6 kinase inhibition assay, and their structural diversity and molecular interactions with key amino acids in the target were considered. This resulted in the identification of two structurally distinct CDK6 inhibitors with IC50 values in the range of 1.48–3.22 μM. The 2 lead compounds both showed H-bonds and solvent exposures similar to the native inhibitor and provide a good platform for lead modification and optimization.
The kinase panel screening of the best hit revealed that it could inhibit CDK6. Despite its inhibitory effect on CDK4, the compound showed good selectivity towards other target kinases. Molecular dynamics studies of these hits suggested that VAL101 was the pivotal residue for stable ligand binding within the pocket, which has been reported in several earlier studies. The potent CDK6 inhibitors screened in this combined methodology validate the feasibility and robustness of combined models and multilayer screening workflows. The structural moieties identified in this study could be further explored by medicinal chemistry for designing specific and potential CDK6 inhibitors.
References
Ghoncheh M, Pournamdar Z, Salehiniya H (2016) Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev 17(S3):43–46. https://doi.org/10.7314/APJCP.2016.17.S3.43
Lim S, Kaldis P (2013) Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140(15):3079–3093. https://doi.org/10.1242/dev.091744
Hou X, Zhang Y, Li W, Hu AJ, Luo C, Zhou W, Hu JK, Daniele SG, Wang J, Sheng JJNC (2018) CDK6 inhibits white to beige fat transition by suppressing RUNX1. Nat Commun 9(1):1023. https://doi.org/10.1038/s41467-018-03451-1
Jia W, Zhao X, Zhao L, Yan H, Li J, Yang H, Huang G, Liu J (2018) Non-canonical roles of PFKFB3 in regulation of cell cycle through binding to CDK4. Oncogene 37(13):1685–1698. https://doi.org/10.1038/s41388-017-0072-4
Wong W (2014) Cycling down glucose concentrations. Sci Signal 7(332):179. https://doi.org/10.1126/scisignal.2005641
Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, Li N, Geng Y, Meng AY, Ordureau AJNCB (2018) G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat Cell Biol 19(3):177–188. https://doi.org/10.1038/ncb3474
Pauklin S, Madrigal P, Bertero A, Vallier LJG (2016) Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev 30(4):421–433. https://doi.org/10.1101/gad.271452.115
O’Leary B, Finn RS, Turner NC (2016) Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13(7):417–430. https://doi.org/10.1038/nrclinonc.2016.26
Zhihong L, Xianghong W, John E, Michael WG, Grace QA, Merrill A (2014) Discovery of AMG 925, a FLT3 and CDK4 dual kinase inhibitor with preferential affinity for the activated state of FLT3. J Med Chem 57(8):3430–3449. https://doi.org/10.1021/jm500118j
Cai BQ, Jin H-x, Yan X-j, Zhu P, Hu G-x (2014) 3D-QSAR and 3D-QSSR studies of thieno[2,3-d]pyrimidin-4-yl hydrazone analogues as CDK4 inhibitors by CoMFA analysis. Acta Pharmacol Sin 35(1):151–160. https://doi.org/10.1038/aps.2013.105
Dong K, Yang X, Zhao T, Zhu X (2017) An insight into the inhibitory selectivity of 4-(Pyrazol- 4-yl)-pyrimidines to CDK4 over CDK2. Mol Simul 43(8):599–609. https://doi.org/10.1080/08927022.2017.1279283
Tadesse S, Yu M, Mekonnen LB, Lam F, Islam S, Tomusange K, Rahaman MH, Noll B, Basnet SKC, Teo T (2017) Highly potent, selective, and orally bioavailable 4-Thiazol-N-(pyridin-2-yl)pyrimidin-2-amine cyclin-dependent kinases 4 and 6 inhibitors as anticancer drug candidates: design, synthesis, and evaluation. J Med Chem 60(5):1892–1915. https://doi.org/10.1021/acs.jmedchem.6b01670
Mahale S, Bharate SB, Manda S, Joshi P, Bharate SS, Jenkins PR, Vishwakarma RA, Chaudhuri B (2014) Biphenyl-4-carboxylic acid [2-(1H-Indol-3-yl)-ethyl]-methylamide (CA224), a nonplanar analogue of fascaplysin, inhibits Cdk4 and tubulin polymerization: evaluation of in vitro and in vivo anticancer activity. J Med Chem 57(22):9658–9672. https://doi.org/10.1021/jm5014743
Tsou HR, Otteng M, Tran T, Floyd MJB, Rabindran SK (2008) 4-(Phenylaminomethylene)isoquinoline-1,3(2 H,4 H)-diones as potent and selective inhibitors of the cyclin-dependent kinase 4 (CDK4). J Med Chem 51(12):3507–3525. https://doi.org/10.1021/jm800072z
Tsou HR, Liu X, Birnberg G, Kaplan J, Otteng M, Tran T, Kutterer K, Tang Z, Suayan R, Zask A (2009) Discovery of 4-(Benzylaminomethylene)isoquinoline-1,3-(2H,4H)-diones and 4-[(Pyridylmethyl)aminomethylene]isoquinoline-1,3-(2H,4H)-diones as potent and selective inhibitors of the cyclin-dependent kinase 4. J Med Chem 52(8):2289–2310. https://doi.org/10.1021/jm801026e
Shivakumar D, Harder E, Damm W, Friesner RA, Sherman W (2012) Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J Chem Theory Comput 8(8):2553–2558. https://doi.org/10.1021/ct300203w
Halgren AT (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49(2):377–389. https://doi.org/10.1021/ci800324m
Nayal M, Honig B (2006) On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins-Struct Funct Bioinform 63(4):892–906. https://doi.org/10.1002/prot.20897
Dixon SL, Smondyrev AM, Rao SN (2006) PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des 67(5):370–372. https://doi.org/10.1111/j.1747-0285.2006.00384.x
Truchon J-F, Bayly CI (2007) Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J Chem Inf Model 47(2):488–508. https://doi.org/10.1021/ci600426e
Cho YS, Angove H, Brain C, Chen CHT, Cheng H, Cheng R, Chopra R, Chung K, Congreve M, Dagostin C, Davis DJ, Felten R, Giraldes J, Hiscock SD, Kim S, Kovats S, Lagu B, Lewry K, Loo A, Lu Y, Luzzio M, Maniara W, McMenamin R, Mortenson PN, Benning R, O’Reilly M, Rees DC, Shen J, Smith T, Wang Y, Williams G, Wrona W, Xu M, Yang F, Howard S (2012) Fragment-based discovery of 7-Azabenzimidazoles as potent, highly selective, and orally active CDK4/6 inhibitors. ACS Med Chem Lett 3(6):445–449. https://doi.org/10.1021/ml200241a
Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, Bergqvist S, Solowiej J, Diehl W, He YA (2016) Spectrum and degree of CDK drug interactions predicts clinical performance. Mol Cancer Ther 15(10):2273–2281. https://doi.org/10.1158/1535-7163.MCT-16-0300
Sakthivel S, Habeeb SKM (2018) Combined pharmacophore, virtual screening and molecular dynamics studies to identify Bruton’s tyrosine kinase inhibitors. J Biomol Struct Dyn 36(16):4320–4337. https://doi.org/10.1080/07391102.2017.1415821
Hu Y, Zhou L, Zhu X, Dai D, Bao Y, Qiu Y (2019) Pharmacophore modeling, multiple docking, and molecular dynamics studies on Wee1 kinase inhibitors. J Biomol Struct Dyn 37(10):2703–2715. https://doi.org/10.1080/07391102.2018.1495576
Saavedra LM, Romanelli GP, Duchowicz PR (2018) Quantitative structure-activity relationship (QSAR) analysis of plant-derived compounds with larvicidal activity against Zika Aedes aegypti (Diptera: Culicidae) vector using freely available descriptors. Pest Manag Sci 74(7):1608–1615. https://doi.org/10.1002/ps.4850
Di L, Fish PV, Mano T (2012) Bridging solubility between drug discovery and development. Drug Discov Today 17(9–10):486–495. https://doi.org/10.1016/j.drudis.2011.11.007
Carlson HA (2002) Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol 6(4):447–452. https://doi.org/10.1016/S1367-5931(02)00341-1
Sherman W, Day T, Jacobson MP, Friesner RA, Farid R (2006) Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49(2):534–553. https://doi.org/10.1021/jm050540c
Godschalk F, Genheden S, Soderhjelm P, Ryde U (2013) Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Phys Chem Chem Phys 15(20):7731–7739. https://doi.org/10.1039/C3CP00116D
Karplus M, McCammon JA (2002) Molecular dynamics simulations of biomolecules. Nat Struct Biol 9(9):646–652. https://doi.org/10.1021/ar020082r
Hollingsworth SA, Dror RO (2018) Molecular dynamics simulation for all. Neuron 99(6):1129–1143. https://doi.org/10.1016/j.neuron.2018.08.011
Makarov GI, Makarova TM, Sumbatyan NV, Bogdanov AA (2016) Investigation of ribosomes using molecular dynamics simulation methods. Biochemistry 81(13):1579–1588. https://doi.org/10.1134/S0006297916130010
Teli MK, Rajanikant GK (2012) Pharmacophore generation and atom-based 3D-QSAR of novel quinoline-3-carbonitrile derivatives as Tpl2 kinase inhibitors. J Enzyme Inhib Med Chem 27(4):558–570. https://doi.org/10.3109/14756366.2011.603128
Cho YS, Borland M, Brain C, Chen CHT, Cheng H, Chopra R, Chung K, Groarke J, He G, Hou Y, Kim S, Kovats S, Lu Y, O’Reilly M, Shen J, Smith T, Trakshel G, Vögtle M, Xu M, Xu M, Sung MJ (2010) 4-(Pyrazol-4-yl)-pyrimidines as selective inhibitors of cyclin-dependent kinase 4/6. J Med Chem 53(22):7938–7957. https://doi.org/10.1021/ml200241a
Lyne PD, Lamb ML, Saeh JC (2006) Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem 49(16):4805–4808. https://doi.org/10.1021/jm060522a
Acknowledgements
We thank West China School of Pharmacy Sichuan University for providing Schrödinger software and technical support for this study.
Funding
This research was funded by Sichuan Applied Basic Research Project (No. 2019YJ0108).
Author information
Authors and Affiliations
Contributions
The authors’ contributions to the manuscript titled “Discovery of new small-molecule cyclin-dependent kinase 6 inhibitors through computational approaches” are listed as follows: XL contributed to Writing-Original Draft, Conceptualization, Methodology. Yu Zhao contributed to Methodology, Verification, Writing-Review & Editing. PT contributed to Methodology, Verification and Formal analysis. XD contributed to Investigation and Verification. RL contributed to Supervision, Methodology and Formal analysis. QW contributed to Writing-Review & Editing. FL contributed to Writing-Review & Editing. JH contributed to Funding acquisition, Conceptualization, Resources, Writing-Review & editing. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Declaration of interest statement the authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Luo, X., Zhao, Y., Tang, P. et al. Discovery of new small-molecule cyclin-dependent kinase 6 inhibitors through computational approaches. Mol Divers 25, 367–382 (2021). https://doi.org/10.1007/s11030-020-10120-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-020-10120-3