
Abstract
Maple Syrup Urine Disease (MSUD) is an autosomal recessive inborn error of metabolism (IEM), responsible for the accumulation of the branched-chain amino acids (BCAA) leucine, isoleucine, and valine, in addition to their α-keto acids α-ketoisocaproic acid (KIC), α-keto-β-methylvaleric acid (KMV), and α-ketoisovaleric acid (KIV) in the plasma and urine of patients. This process occurs due to a partial or total blockage of the dehydrogenase enzyme activity of branched-chain α-keto acids. Oxidative stress and inflammation are conditions commonly observed on IEM, and the inflammatory response may play an essential role in the pathophysiology of MSUD. We aimed to investigate the acute effect of intracerebroventricular (ICV) administration of KIC on inflammatory parameters in young Wistar rats. For this, sixteen 30-day-old male Wistar rats receive ICV microinjection with 8 µmol KIC. Sixty minutes later, the animals were euthanized, and the cerebral cortex, hippocampus, and striatum structures were collected to assess the levels of pro-inflammatory cytokines (INF-γ; TNF-α, IL-1β). The acute ICV administration of KIC increased INF-γ levels in the cerebral cortex and reduced the levels of INF-γ and TNF-α in the hippocampus. There was no difference in IL-1β levels. KIC was related to changes in the levels of pro-inflammatory cytokines in the brain of rats. However, the inflammatory mechanisms involved in MSUD are poorly understood. Thus, studies that aim to unravel the neuroinflammation in this pathology are essential to understand the pathophysiology of this IEM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inborn errors of metabolism (IEMs) are caused by genetic mutations that impair protein or enzyme synthesis (Wasim et al. 2018). Maple syrup urine disease (MSUD) is a disorder that affects the metabolism of amino acids (Menkes 1959; Dancis et al. 1972; Chuang et al. 2008). In this particular IEM, there is the accumulation of leucine, isoleucine, and valine, as well as the transaminated branched-chain α-ketoacids α-ketoisocaproic acid (KIC), α-ketoisovaleric acid (KIV), and α-keto-β-methylvaleric acid (KMV), in body tissues and fluids (Chuang and Shih 2001).
The estimated worldwide frequency of MSUD is approximately 1:185,000 newborns (Chuang and Shih 2001). In the first weeks of life, patients with MSUD can present lethargy, hypoglycemia, ketoacidosis, apnea, and ataxia symptoms (Chuang and Shih 2001; Schonberger 2004). Furthermore, they may demonstrate psychomotor and mental retardation, cerebral hemisphere atrophy, generalized cerebral edema, delayed myelination, spongy degeneration of the cerebral white matter, and encephalopathy that can result in irreversible coma and death (Treacy et al. 1992; Chuang and Shih 2001; Berry et al. 2003; Jain et al. 2013; Klee et al. 2013).
Patients affected by MSUD also present several neurological problems, including learning and memory deficits, caused by alterations in the synthesis of glutamate. In turn, changes in glutamate levels can occur due to disturbances in the metabolism of branched-chain amino acids (BCAAs) (Scaini et al. 2017). Studies have demonstrated an association between increased levels of leucine and/or KIC and neurological symptoms, suggesting that these may be the major neurotoxic components in MSUD (Snyderman et al. 1964; Chuang and Shih 2001). Furthermore, the accumulation of metabolites in the brain promotes mitochondrial dysfunction, resulting in deficits in brain energy metabolism (Sgaravatti et al. 2003; Ribeiro et al. 2008), oxidative stress, reduced antioxidant defenses, decreased levels of neurotrophins (Bridi et al. 2005; Funchal et al. 2006; Mescka et al. 2011; Sitta et al. 2014; Wisniewski et al. 2016; Taschetto et al. 2017). In addition, it also reduces the brain uptake of essential amino acids, culminating in changes in neurotransmitter concentrations (Wajner et al. 2000; Tavares et al. 2000; Ronald Zielke et al. 2002). In a previous study conducted by our group, evaluating the acute and chronic administration of KIC in different parameters, we observed similar damage between the acute and the chronic administration of KIC and even more significant damage in the acute administration in some parameters of oxidative stress (Taschetto et al. 2017).
Several biological mechanisms can trigger neuroinflammation, including oxidative stress, microglial reactions, and exacerbated release of inflammatory mediators such as cytokines, chemokines, prostaglandins, complement cascade proteins, and ROS, which play a crucial role in the development of neurodegenerative diseases (Niranjan 2013; Vivekanantham et al. 2015; Gelders et al. 2018). In addition, previous studies have demonstrated that the inflammatory response plays an essential role in the pathogenesis of some IEMs, including phenylketonuria (Deon et al. 2015), type I glutaric acidemia (Seminotti et al. 2016), and methylmalonic acidemia (Ribeiro et al. 2013).
According to Mescka et al. (2015a, b), MSUD patients on a protein-restricted diet have high levels of pro-inflammatory cytokines. Furthermore, animal models and cell cultures studies have shown that BCAAs alter the balance between pro- and anti-inflammatory cytokines (De Simone et al. 2013; Rosa et al. 2016). Therefore, considering the hypothesis that KIC can cause central nervous system (CSN) dysfunction in MSUD, this study aimed to evaluate the acute effect of intracerebroventricular (ICV) administration of KIC on inflammatory parameters in the cerebral cortex, hippocampus, and striatum of young Wistar rats. Since there are few studies on inflammation in this disease, the present study may contribute to a better understanding of the pathophysiology of MSUD.
Materials and methods
Animals
This study used male Wistar rats (60–80 g) with 30 days of life from the Central Animal House of the Universidade do Extremo Sul Catarinense (UNESC). Rats were caged in 2 groups of 8 animals with free access to food and water, maintenance of a 12-hour light/dark cycle (lights on at 7:00 am and off at 7:00 pm), and at a temperature of 23 ± 1 °C. All experimental procedures were approved by the UNESC ethics committee (protocol number 23/2021) and followed the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Brazilian Society of Neurosciences and Behavior for animal care.
Administration of α-ketoisocaproic acid (KIC)
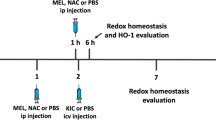
The animals were anesthetized intramuscularly with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg) and then placed in the stereotaxic apparatus. A 2 µL solution of 8 µmol KIC (Taschetto et al. 2017) was dissolved in freshly prepared artificial cerebrospinal fluid (ACSF) (NaCl 147 mM; KCl 2.9 mM; MgCl2 1.6 mM; CaCl2 1, 7 mM and 2.2 mM dextrose). An ACSF-only solution with the same volume and concentration was used for the control group. A small hole was drilled in the skull for microinjection, and the respective solution was slowly injected bilaterally over 4 min into the lateral ventricles through a needle connected by a polyethylene tube to a 10 µL Hamilton syringe (de Castro Vasques et al. 2004). The needle was left in place for an additional 1 min before being gently removed. The pH of each solution was previously adjusted to 7.4 with 0.1 N NaOH or 0.1 N HCl. The injection coordinates used were 0.6 mm posterior to bregma, 1.0 mm lateral to the midline, and 3.2 mm ventral of the dura mater (Paxinos and Watson 1986).
Euthanasia and sample preparation
After 60 min of the ICV administration of KIC or ACSF, the rats were euthanized by guillotine. Then, the brain was quickly excised in a Petri dish and placed on an ice plate, where the cerebral cortex, hippocampus, and striatum were dissected and stored at -80º for further analysis. For ELISA measurement, 100 mg of the sample was homogenized in 1 mL of PBS, centrifuged at 300 rmp, 4ºC for 10 min, and the supernatant was used for the analysis.
Inflammation parameters
For measurement of pro-inflammatory cytokines (INF-γ, TNF-α, IL-1β), the enzyme-linked immunosorbent assay (DuoSet ELISA) capture method was used (R&D System, Inc., Minneapolis, USA). ELISA is an enzyme immunoassay in which antigen-antibody reactions are detectable. All steps for this assay were conducted according to the manufacturer’s instructions. First, the plate was prepared according to the steps described below: the capture antibody was diluted and then coated in a microplate that was sealed and incubated overnight; after, a wash buffer process was conducted in the microplate three times, and remaining wash buffer was removed for the addition of reagent diluent in each well; the microplate was again incubated for a minimum of 1 h. This process was done twice at room temperature (R&D System, Inc., Minneapolis, USA).
After the microplate preparation, 100 µL of the sample prepared with the reagent diluent was added to each well, covered, and incubated for 2 h at room temperature. Right after, the aspiration and wash described in plate preparation were conducted again. Then, the detection antibody (100 µL) in reagent diluent was added to each well, covered, incubated for 2 h at room temperature, and aspirated and washed as described. After, a working dilution of Streptavidin-HRP B (100 µL) was added in each well, covered, and incubated for 20 min at room temperature, with the aspiration and wash steps performed again. The same process was made with a substrate solution (100 µL). Finally, 50 µL of stop solution was added to each well and measured spectrophotometrically at 450 nm d (R&D System, Inc., Minneapolis, USA). Each pro-inflammatory cytokine (INF-γ, TNF-α, IL-1β) was dosed separately, according to the ELISA assay technique described above.
Statistical analysis
Data on inflammatory parameters were analyzed using Student’s t-test, with a significance level of 5% (p-value ≤ 0.05), and are expressed as a mean with standard deviation (±). All analyses were performed using the Statistic software version 7.0, and the graphs were constructed using GraphPad Prism version 7.03.
Results
There were increased INF-γ levels in the cerebral cortex (control: 0.10 pg/mg protein ± 0.02 vs. KIC: 0.12 pg/mg protein ± 0.02; p = 0.0246), while in the hippocampus, the levels of INF-γ were reduced compared to the control group (control: 0.21 pg/mg protein ± 0.06 vs. KIC: 0.12 pg/mg protein ± 0.03; p = 0.0003). In the striatum, there was no change in INF-γ levels after the administration of KIC compared to the control group (control: 0.24 pg/mg protein ± 0.07 vs. KIC: 0.25 pg/mg protein ± 0.08; p = 0.8692) (Fig. 1).
Regarding TNF-α level, ICV administration of KIC caused no difference in the cerebral cortex (control: 22.36 pg/mg protein ± 5.44 vs. KIC: 23.74 pg/mg protein ± 8.79; p = 0.7492) and the striatum (control: 61.54 pg/mg protein ± 11.59 vs. 60.29 pg/mg protein ± 14.83; p = 0.8737) compared to the control group. However, a decrease in TNF-α levels in the hippocampus of rats with ICV administration of KIC was observed compared to the controls (control: 39.24 pg/mg protein ± 4.78 vs. KIC: 27.17 pg/mg protein ± 5.85; p = 0.0073) (Fig. 2).
Administration of KIC by ICV did not cause statistically significant differences in IL-1β levels in the cerebral cortex (control: 316.74 pg/mg protein ± 75,72 vs. 333.72 pg/mg protein ± 129,15; p = 0.7868), striatum (control: 675.91 pg/mg protein ± 123.78 vs. KIC: 739.75 pg/mg protein ± 229,27; p = 0.4485), and hippocampus (control: 377.38 pg/mg protein ± 107.10 vs. KIC: 361.60 pg/mg protein ± 72,74; p = 0.7046) compared to the control group (Fig. 3).

The effect of acute intracerebroventricular administration (ICV) of α-ketoisocaproic acid (KIC) on interferon-gamma (INF-γ) levels in the cerebral cortex (A), striatum (B), hippocampus (C) of 30-day-old male rats. Data are expressed as mean with standard deviation (mean ± SD). Student’s t-test (n = 6). *p < 0.05 vs. Control

The effect of acute intracerebroventricular (ICV) administration of α-ketoisocaproic acid (KIC) on tumor necrosis factor-alpha (TNF-α) levels in the cerebral cortex (A), striatum (B), hippocampus (C) of 30-day-old male rats. Data are expressed as mean with standard deviation (mean ± SD). Student’s t-test (n = 6). *p < 0.05 vs. Control

The effect of acute intracerebroventricular (ICV) administration of α-ketoisocaproic acid (KIC) on interleukin-1 beta (IL-1β) levels in the cerebral cortex (A), striatum (B), hippocampus (C) of 30-day-old male rats. Data are expressed as mean with standard deviation (mean ± SD). Student’s t-test (n = 6). *p < 0.05 vs. Control
Discussion
Leucine and its α-keto acids, such as KIC, are potentially neurotoxic in MSUD, especially during metabolic decompensation crises, where this compound concentration dramatically increases (Snyderman et al. 1964; Chuang and Shih 2001). Furthermore, Zinnanti et al. (2008) showed a correlation between severe brain damage and death in mice with classic and intermediate MSUD and high levels of KIC (above 100 µmol) in the animals’ brains. Moreover, acute administration of KIC by ICV was associated with lower levels of BDNF in the hippocampus, striatum, and cerebral cortex and lower levels of neuronal growth factor (NGF) in the hippocampus of young rats (Wisniewski et al. 2016).
Previous studies demonstrate that BCAAs influence immune functions and, in MSUD, also affect the balance of pro- and anti-inflammatory cytokines in the brain of Wistar rats with acute and chronic administration of BCAAs (Calder 2006; Rosa et al. 2016). Inflammation often causes acute decompensation and neurological deterioration in MSUD patients (Chuang and Shih 2001). So, our results indicate that ICV administration of KIC was associated with changes in the levels of pro-inflammatory cytokines INF-γ and TNF-α. There was an increase in INF-γ levels in the cerebral cortex, while a reduction in INF-γ and TNF-α levels was observed in the hippocampus. IL-1β levels did not change.
The findings of Wessler et al. (2019) also related the pathogenesis of MSUD with a pro-inflammatory state in an animal model. Different from our results, acute and subcutaneous administration of BCAA and BCAA together with lipopolysaccharide (LPS) was associated with increased IL-1β levels in the cerebral cortex. The same administration of BCAA and LPS was related to higher levels of TNF-α in the cerebral cortex and hippocampus and of INF-γ only in the hippocampus (Wessler et al. 2019; Rosa et al. 2016) found an increase in IL-1β and TNF-α levels in the cerebral cortex of Wistar rats that received acute subcutaneous administration of BCAA, compared to the control group. On the other hand, animals that received chronic subcutaneous administration of BCAA presented a decrease of IL-1β in the cerebral cortex and a reduction in the levels of INF-γ in the cerebral cortex and hippocampus compared to the controls (Rosa et al. 2016; Scaini et al. 2018), in a study with 12 MSUD patients treated with a protein-restricted diet with isoleucine and valine supplementation, observed elevated levels of pro-inflammatory cytokines, including IFN-γ, TNF-α, IL-1β, and IL-6 when compared to a control group. These results highlight the disparities in the markers of neuroinflammation existing in MSUD.
The levels of inflammatory mediators can change according to the brain structure analyzed due to receptors and cell populations. Thus, it is impossible to establish the same inflammatory pattern in different brain areas (Arisi 2014). When an imbalance between pro- and anti-inflammatory activities occurs and brain homeostasis is disturbed, neuronal compromise can ensue. During immune activation, the increase in pro-inflammatory cytokines can induce the production of oxidants, prostaglandins, and reactive species, exacerbating inflammation (Muralidharan and Mandrekar 2013). Chronic neuroinflammation can occur when the release of pro-inflammatory factors is continuous and persistent, as in some neurodegenerative and neurological diseases (Di Benedetto et al. 2017), including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, as well as multiple sclerosis (Stephenson et al. 2018).
There is an increase in TNF-α, IFN-ɣ, IL-1β, and IL-6 in MSUD (Amaral and Wajner 2022). However, studies with biomarkers are still conflicting. Cytokines have the potential to exacerbate or reduce neuroinflammation. IL-1β and TNF-α are essential in inflammation and neurodegenerative disease progression (Opal and DePalo 2000). Likewise, IFN-ɣ is produced mainly by T helper type 1 (Th1) lymphocytes, and during the Th1 response, IFN-ɣ can induce high production of reactive species (Mescka et al. 2015a).
In the case of TNF-α, its presence is responsible for activating nuclear factor kappa B (NFκB), neural cell apoptosis, and c-Jun N-terminal kinase (JNK), pathways and mechanisms involved in neuroinflammatory status (Olmos and Lladó 2014; Muhammad 2020). In turn, T cell release IFN-ɣ in CNS, which is linked to the activation of the production of TNF-α by microglia. IFN-ɣ is also directly connected with neuroinflammation, although its mechanisms are not entirely described (Olmos and Lladó 2014). The outcomes of these processes are various: glutamate exocytosis, an increase of excitatory synaptic, a decrease in the expression of GABA receptors, neuronal death, and oxidative stress (Olmos and Lladó 2014).
Curiously, in our study, lower levels of TNF-α and IFN-ɣ were found in the hippocampus of rats with acute ICV administration of KIC. It is well known that microglia is essential for the inflammation in the brain and that its dysfunction can affect this process (Colonna and Butovsky 2017; Teleanu et al. 2022). Usually, there is a consensus that the activation of macrophages M1 is pro-inflammatory and induces secretion of cytokines like TNF-α (whose production is also related to IFN-ɣ levels) and IL-1β, while the activation of macrophages M2 is anti-inflammatory (Varella and Forte 2001; Colonna and Butovsky 2017). However, the literature has observed that the activity of macrophages M1 or M2 is context dependable, especially in neurodegeneration (Colonna and Butovsky 2017). For example, in neurodegenerative diseases like amyotrophic lateral sclerosis, the gene expression of macrophages does not fit in the M1/M2 normal activation, reflecting the microglial response from the protein aggregation, oxidative stress and cellular death (Colonna and Butovsky 2017).
It is hypothesized that the levels of BCAA in the organism might affect the functional activity of microglia, influencing the M1/M2 activity regulation. According to that, the study of de Simone et al. (2013), with primary glial cultures from the cortex of 1-day-old rats, demonstrated that high levels of BCAA reduced transcripts encoding levels for M1 genes. That may indicate that high levels of BCAA can change the immune proprieties of microglia, generating a lower expression of genes M1 and, consequently, a reduced secretion of IL-1β and TNF-α. This less inflammatory M1 response suggests a microglial activity less efficient for the damage (De Simone et al. 2013). So, it is possible to assume that KIC levels could have created an inadequate pro-inflammatory response from macrophages M1, causing lower levels of IFN-ɣ and, consequently, of TNF-α to be secreted on the animals’ hippocampus, but not sufficient to affect IL-1β levels significantly.
Studies have linked inflammation as a risk factor for the development of disorders in the balance between reactive species production and antioxidant defenses. Taschetto et al. (2017) showed damage in the hippocampus, striatum, and cerebral cortex, increased oxidative stress, changes in enzymatic antioxidant defenses, and behavioral changes in Wistar rats after 1 h and 15 days of ICV administration of KIC. These results may also be related to a recent study conducted by Farias et al. (2021), where was demonstrate that ICV administration of KIC reduced the activity of mitochondrial complexes in vivo and in vitro, at the same time that induced a high production of reactive species and was able to alter cell viability, showing the toxicity of KIC. These findings corroborate with the neuroinflammatory conditions in MSUD once it is indicated that dysfunctions in the respiratory chair can generate high concentrations of reactive species, which, in turn, can induce neuroinflammation, establishing a significant connection between inflammation and oxidative stress. This sets a vicious cycle that is capable of perpetuating and propagating the inflammatory response (Lugrin et al. 2014), which could be related to MSUD pathophysiology.
Conclusion
Our study demonstrated that ICV administration of KIC was related to changes in INF-y and TNF-α levels in the cerebral cortex and hippocampus of young male rats. However, decreasing TNF-α and IFN-ɣ levels in the animals’ hippocampus points out some important future considerations. Despite these findings supporting the hypothesis that immunological mechanisms are involved in MSUD pathogenesis, especially neuroinflammation, they highlight the need to clarify the activation conformation of macrophages M1 and M2 in this disease since it may represent a key point for an insufficient pro-inflammatory response, as found here. Thus, considering the few studies and the disparity of results on neuroinflammation in MSUD, as well as the possible pathways with influence on the topic, more studies are needed to understand the pathophysiology of this disease better.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available due to ethics reasons but are available from the corresponding author on reasonable request.
References
Amaral AU, Wajner M (2022) Pathophysiology of maple syrup urine disease: focus on the neurotoxic role of the accumulated branched-chain amino acids and branched-chain α-keto acids. Neurochem Int 157:105360. https://doi.org/10.1016/j.neuint.2022.105360
Arisi GM (2014) Nervous and immune systems signals and connections: cytokines in hippocampus physiology and pathology. Epilepsy Behav 38:43-47.?https://doi.org/10.1016/j.yebeh.2014.01017.017
Berry GT, Kaplan PB, Kaye EM et al (2003) MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology 45:393–399. https://doi.org/10.1007/s00234-003-0955-7
Bridi R, Braun CA, Zorzi GK et al (2005) α-Keto acids accumulating in maple syrup urine Disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab Brain Dis 20:155–167. https://doi.org/10.1007/s11011-005-4152-8
Calder PC (2006) Branched-chain amino acids and immunity. J Nutr 136. https://doi.org/10.1093/jn/136.1.288S. :288S-293S
Chuang D, Shih V (2001) Maple syrup urine disease (branched-chain ketoaciduria). The metabolic and molecular bases of inherited disease. McGrawHill, New York
Chuang D, wYNN M, Shih V (2008) Doença da urina do xarope de bordo (cetoacidúria de cadeia ramificada). As bases metabólicas e moleculares da doença hereditária. McGrawHill, Nova York, pp 1971–2005
Colonna M, Butovsky O (2017) Microglia function in the Central Nervous System during Health and Neurodegeneration. Annu Rev Immunol 35:441–468. https://doi.org/10.1146/annurev-immunol-051116-052358
Dancis J, Hutzler J, Snyderman SE, Cox RP (1972) Enzyme activity in classical and variant forms of maple syrup urine disease. J Pediatr 81:312–320. https://doi.org/10.1016/S0022-3476(72)80301-9
de Castro Vasques V, Avila de Boer M, Diligenti F et al (2004) Intrahippocampal administration of the α-keto acids accumulating in maple syrup urine disease provokes learning deficits in rats. Pharmacol Biochem Behav 77:183–190. https://doi.org/10.1016/j.pbb.2003.10.013
De Simone R, Vissicchio F, Mingarelli C et al (2013) Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim et Biophys Acta (BBA) - Mol Basis Disease 1832:650–659. https://doi.org/10.1016/j.bbadis.2013.02.001
Deon M, Sitta A, Faverzani JL et al (2015) Urinary biomarkers of oxidative stress and plasmatic inflammatory profile in phenylketonuric treated patients. Int j dev neurosci 47:259–265. https://doi.org/10.1016/j.ijdevneu.2015.10.001
Di Benedetto S, Müller L, Wenger E et al (2017) Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehavioral Reviews 75:114–128. https://doi.org/10.1016/j.neubiorev.2017.01.044
Farias HR, Gabriel JR, Cecconi ML et al (2021) The metabolic effect of α-ketoisocaproic acid: in vivo and in vitro studies. Metab Brain Dis 36:185–192. https://doi.org/10.1007/s11011-020-00626-y
Funchal C, Latini A, Jacques-Silva MC et al (2006) Morphological alterations and induction of oxidative stress in glial cells caused by the branched-chain α-keto acids accumulating in maple syrup urine disease. Neurochem Int 49:640–650. https://doi.org/10.1016/j.neuint.2006.05.007
Gelders G, Baekelandt V, Van der Perren A (2018) Linking neuroinflammation and neurodegeneration in Parkinson’s Disease. J Immunol Res 2018:1–12. https://doi.org/10.1155/2018/4784268
Jain P, Sharma S, Sankhyan N et al (2013) Imaging in neonatal maple syrup urine disease. Indian J Pediatr 80:87–88. https://doi.org/10.1007/s12098-012-0850-5
Klee D, Thimm E, Wittsack HJ et al (2013) Structural white matter changes in adolescents and young adults with maple syrup urine disease. J Inherit Metab Dis 36:945–953. https://doi.org/10.1007/s10545-012-9582-y
Lugrin J, Rosenblatt-Velin N, Parapanov R, Liaudet L (2014) The role of oxidative stress during inflammatory processes. Biol Chem 395:203–230. https://doi.org/10.1515/hsz-2013-0241
Menkes JH (1959) Maple syrup disease; isolation and identification of organic acids in the urine. Pediatrics 23:348–353
Mescka C, Moraes T, Rosa A et al (2011) In vivo neuroprotective effect of L-carnitine against oxidative stress in maple syrup urine disease. Metab Brain Dis 26:21–28. https://doi.org/10.1007/s11011-011-9238-x
Mescka CP, Guerreiro G, Donida B et al (2015a) Investigation of inflammatory profile in MSUD patients: benefit of L-carnitine supplementation. Metab Brain Dis 30:1167–1174. https://doi.org/10.1007/s11011-015-9686-9
Mescka CP, Guerreiro G, Hammerschmidt T et al (2015b) l-Carnitine supplementation decreases DNA damage in treated MSUD patients. Mutat Research/Fundamental Mol Mech Mutagen 775:43–47. https://doi.org/10.1016/j.mrfmmm.2015.03.008
Muhammad M (2020) Tumor Necrosis Factor Alpha: A Major Cytokine of Brain Neuroinflammation. In: Behzadi P (ed) Cytokines. IntechOpen
Muralidharan S, Mandrekar P (2013) Cellular stress response and innate immune signaling: integrating pathways in host defense and inflammation. J Leukoc Biol 94:1167–1184. https://doi.org/10.1189/jlb.0313153
Niranjan R (2013) Molecular basis of Etiological Implications in Alzheimer’s Disease: Focus on Neuroinflammation. Mol Neurobiol 48:412–428. https://doi.org/10.1007/s12035-013-8428-4
Olmos G, Lladó J (2014) Tumor necrosis factor alpha: a link between Neuroinflammation and Excitotoxicity. Mediat Inflamm 2014:1–12. https://doi.org/10.1155/2014/861231
Opal SM, DePalo VA (2000) Anti-inflammatory cytokines. Chest 117:1162–1172. https://doi.org/10.1378/chest.117.4.1162
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press, Sydney
Ribeiro CA, Sgaravatti ÂM, Rosa RB et al (2008) Inhibition of Brain Energy metabolism by the branched-chain amino acids accumulating in maple syrup urine disease. Neurochem Res 33:114–124. https://doi.org/10.1007/s11064-007-9423-9
Ribeiro LR, Della-Pace ID, de Oliveira Ferreira AP et al (2013) Chronic administration of methylmalonate on young rats alters neuroinflammatory markers and spatial memory. Immunobiology 218:1175–1183. https://doi.org/10.1016/j.imbio.2013.04.008
Ronald Zielke H, Zielke CL, Baab PJ, Collins RM (2002) Large neutral amino acids auto exchange when infused by microdialysis into the rat brain: implication for maple syrup urine disease and phenylketonuria. Neurochem Int 40:347–354. https://doi.org/10.1016/S0197-0186(01)00077-8
Rosa L, Scaini G, Furlanetto CB et al (2016) Administration of branched-chain amino acids alters the balance between pro‐inflammatory and anti‐inflammatory cytokines. Int j dev neurosci 48:24–30. https://doi.org/10.1016/j.ijdevneu.2015.11.002
Scaini G, Tonon T, de Souza CFM et al (2017) Serum markers of neurodegeneration in maple syrup urine disease. Mol Neurobiol 54:5709–5719. https://doi.org/10.1007/s12035-016-0116-8
Scaini G, Tonon T, Moura de Souza CF et al (2018) Evaluation of plasma biomarkers of inflammation in patients with maple syrup urine disease. J Inherit Metab Dis 41:631–640. https://doi.org/10.1007/s10545-018-0188-x
Schonberger S (2004) Dysmyelination in the brain of adolescents and young adults with maple syrup urine disease. Mol Genet Metab 82:69–75. https://doi.org/10.1016/j.ymgme.2004.01.016
Seminotti B, Amaral AU, Ribeiro RT et al (2016) Oxidative stress, disrupted Energy Metabolism, and altered signaling pathways in Glutaryl-CoA dehydrogenase knockout mice: potential implications of quinolinic acid toxicity in the neuropathology of Glutaric Acidemia Type I. Mol Neurobiol 53:6459–6475. https://doi.org/10.1007/s12035-015-9548-9
Sgaravatti AM, Rosa RB, Schuck PF et al (2003) Inhibition of brain energy metabolism by the α-keto acids accumulating in maple syrup urine disease. Biochimica et Biophysica Acta (BBA) -. Mol Basis Disease 1639:232–238. https://doi.org/10.1016/j.bbadis.2003.09.010
Sitta A, Ribas GS, Mescka CP et al (2014) Neurological damage in MSUD: the role of oxidative stress. Cell Mol Neurobiol 34:157–165. https://doi.org/10.1007/s10571-013-0002-0
Snyderman SE, Norton PM, Roitman E, Holt LE (1964) MAPLE SYRUP URINE DISEASE, WITH PARTICULAR REFERENCE TO DIETOTHERAPY. Pediatrics 34:454–472
Stephenson J, Nutma E, van der Valk P, Amor S (2018) Inflammation in CNS neurodegenerative diseases. Immunology 154:204–219. https://doi.org/10.1111/imm.12922
Taschetto L, Scaini G, Zapelini HG et al (2017) Acute and long-term effects of intracerebroventricular administration of α-ketoisocaproic acid on oxidative stress parameters and cognitive and noncognitive behaviors. Metab Brain Dis 32:1507–1518. https://doi.org/10.1007/s11011-017-0035-z
Tavares RG, Santos CES, Tasca CI et al (2000) Inhibition of glutamate uptake into synaptic vesicles of rat brain by the metabolites accumulating in maple syrup urine disease. J Neurol Sci 181:44–49. https://doi.org/10.1016/S0022-510X(00)00402-0
Teleanu DM, Niculescu A-G, Lungu II et al (2022) An overview of oxidative stress, Neuroinflammation, and neurodegenerative Diseases. IJMS 23:5938. https://doi.org/10.3390/ijms23115938
Treacy E, Clow CL, Reade TR et al (1992) Maple syrup urine disease: interrelations between branched-chain amino‐, oxo‐ and hydroxyacids; implications for treatment; associations with CNS dysmyelination. J of Inher Metab Disea 15:121–135. https://doi.org/10.1007/BF01800354
Varella PPV, Forte WCN (2001) Citokines: a review. Rev bras alergia imunopatol 24:146–154
Vivekanantham S, Shah S, Dewji R et al (2015) Neuroinflammation in Parkinson’s disease: role in neurodegeneration and tissue repair. Int J Neurosci 125:717–725. https://doi.org/10.3109/00207454.2014.982795
Wajner M, Coelho DM, Barschak AG et al (2000) Reduction of large neutral amino acid concentrations in plasma and CSF of patients with maple syrup urine disease during crises. J Inherit Metab Dis 23:505–512. https://doi.org/10.1023/A:1005668431926
Wasim M, Awan FR, Khan HN et al (2018) Aminoacidopathies: prevalence, etiology, screening, and Treatment Options. Biochem Genet 56:7–21. https://doi.org/10.1007/s10528-017-9825-6
Wessler LB, Miranda Ramos V, Bittencourt Pasquali MA et al (2019) Administration of branched-chain amino acids increases the susceptibility to lipopolysaccharide‐induced inflammation in young Wistar rats. Int j dev neurosci 78:210–214. https://doi.org/10.1016/j.ijdevneu.2019.07.007
Wisniewski MSW, Carvalho-Silva M, Gomes LM et al (2016) Intracerebroventricular administration of α-ketoisocaproic acid decreases brain-derived neurotrophic factor and nerve growth factor levels in brain of young rats. Metab Brain Dis 31:377–383. https://doi.org/10.1007/s11011-015-9768-8
Zinnanti WJ, Lazovic J, Griffin K et al (2008) Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain 132:903–918. https://doi.org/10.1093/brain/awp024
Acknowledgements
This research was supported by grants from Universidade do Extremo Sul Catarinense (UNESC), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina.
Funding
This research was supported by grants from Universidade do Extremo Sul Catarinense (UNESC), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina.
Author information
Authors and Affiliations
Contributions
FR and ELS developed the study conception and design. ISL, CPDT, DDC and MLSF performed the experiment. IRL, MM, PCLS and FDP carried out the data analysis. FR, ISL and ELS developed the statistical analysis. The first draft of the manuscript was written by FR and MRQ. JSG and ELS conducted the written review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
None.
Ethics approval
All experimental procedures were approved by the UNESC ethics committee (protocol number 23/2021), following the recommend dations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Brazilian Society of Neurosciences and Behavior for animal care.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rabelo, F., Lemos, I.d.S., Dal Toé, C.P. et al. Acute effects of intracerebroventricular administration of α-ketoisocaproic acid in young rats on inflammatory parameters. Metab Brain Dis 38, 1573–1579 (2023). https://doi.org/10.1007/s11011-023-01193-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01193-8