
Abstract
Significant strides have been made in our understanding of the immune system and its role in cardiac transplant rejection. Despite the growing knowledge of immune responses, the mortality rate following cardiac transplantation remains grim. Related to procedural and pathological complications, toll-like receptor (TLR) and damage-associated molecular pattern (DAMP) signaling is the most direct and earliest interface between tissue integration and the innate immune response. This in turn can activate an adaptive immune response that further damages myocardial tissue. Furthermore, relevant literature on the status of DAMPs in the context of heart-transplantation remains limited, warranting further attention in clinical and translational research. This review aims to critically appraise the perspectives, advances, and challenges on DAMP-mediated innate immune response in the immune-mediated rejection of cardiac transplantation. Detailed analysis of the influence of TLR and DAMP signaling in mounting the immune response against the transplanted heart holds promise for improving outcomes through early detection and prevention of varied forms of organ rejection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
About 17.9 million people die from cardiovascular disease (CVD) each year, and in the United States (US), a death from CVD occurs every 36 s on average [1, 2]. Clinically, CVD is an umbrella term for a multitude of diseases of blood vessels, including coronary heart disease (CHD), heart failure, and myocardial infarction (MI). Currently, CVD remains prevalent worldwide. Heidenreich et al. project that about 40% of the US population are at the higher of CVD by 2030 [3]. Interestingly, between 2000 and 2012, the 5-year survival improved from 41 to 48.2% in the United Kingdom; similar to US [4]. Although survival has improved, less than half of patients with newly diagnosed heart disease survive past the 5-year mark.
Heart transplantation (HT) has been considered as a last resort in end-stage HF. Transplant outcomes have been continuously improved by advances in surgical technique and immunosuppressive therapies [5]. Although HT is a life-saving procedure for HF patients, its long-term outcomes remain grim. Immunosuppressive treatment succeeded to increase the 1-year survival rate to greater than 80% and the 5-year survival rate to about 69% [6]. Most long-term complications arise out of infectious diseases, the side effects of immunosuppressive treatments, and progressive cardiac allograft vasculopathy (CAV) in immunocompromised patients [7]. The relevance of the immune response is underlined by these complications and the contemporary advances in transplant survival.
Antibody-mediated rejection (AMR) is the most studied and best described rejection pathway following transplantation. Generally, AMR occurs in approximately 15% of HT recipients, making it the primary cause for cardiac transplant rejection [6]. Symptoms of AMR ranges from mild heart failure to cardiogenic shock [8].
Conventional treatment modalities targeting AMR aim to prevent further immune-mediated injury and provide supportive cardiac treatment, with cardiac re-transplantation reserved for patients who fail in aggressive therapy. Current treatments for AMR consist of plasmapheresis for antibody depletion, immunoadsorption, immunomodulation with intravenous immunoglobulin, and T cell- and B cell-depleting drugs; however, most of these lack long-term control of rejection [9, 10]. Considering the post-transplant survival rate, as well as the obvious limitations in targeting the adaptive immune system, it is critical to identify novel translational avenues.
Damage-associated molecular patterns (DAMPs) are the early signals released from injured cells that stimulate immune responses [11, 12]. Numerous studies of solid-organ transplantation have elucidated the potential role of DAMPs as these danger signals activate and regulate the immune response, yet the mechanisms are poorly defined due to the cross-reactivity of pattern recognition receptors (PRRs) with non-self-materials containing pathogen-associated molecular patterns (PAMPs) [13,14,15] (Table 1). DAMPs are released in perioperative injury during organ procurement and reimplantation and accumulate through organ transplant-specific mechanisms. The accumulation of danger signals is part of the innate immune response which precedes the adaptive immune response and strengthens the signaling mechanisms that constitute transplant rejection. Hence, targeting these mechanisms paves the way to alternate treatment modalities to prevent cardiac allograft rejection. This article critically reviews the advances, perspectives, and challenges of HT with a focus on DAMP-mediated immune response in acute allograft rejection.
Immunosuppressive therapies
Immunosuppressant therapies jumpstarted the modern era of HT in the 1980s leading to the monumental rise in the number of transplant centers and transplanted patients annually [25, 26]. Immunosuppressants have reduced the risk of rejection and minimized the complications. 1-year survival following HT in 1967–1973 was 30%. Between 1982 and 1992, mean survival following HT was increased to 8.5 years due to the introduction of cyclosporine A, a calcineurin inhibitor, which had further increased survival [7, 27]. The median survival rate from 1982 to 2015 was 10.7 years [7]. As of now, immunosuppressive therapies are typically front-loaded immediately after surgery as graft rejection is most common within the first 3–6 months after transplantation [25]. These induction therapies consist of glucocorticoids or glucocorticoid-sparing treatments with agents such as anti-interleukin-2 receptor agonists. The induction dosage is slowly tapered and replaced with long-term maintenance therapy consisting of a triple-drug regimen of prednisone, mycophenolate mofetil, and tacrolimus within hours following surgery [28, 29]. Although this regimen does not improve long-term survival, it reduces the risk of acute rejection [25].
Proliferation signal inhibitors (PSIs) have been used in patients with renal insufficiency, CAV, or malignancies to attenuate or reverse the disease processes [25]. Sirolimus and everolimus are used to inhibit T cell, B cell, and vascular smooth muscle cell proliferation. When used alongside cyclosporine and prednisone, these drugs help prevent acute rejection and slow the progression of CAV in de novo transplant recipients [25, 30]. For patients with chronic CAV, re-transplantation may be an option [30].
Multiple studies have indicated that DAMPs can be targeted therapeutically and that suppressing/inhibiting DAMPs (SAMPs) can be used as candidate drugs [31,32,33,34,35]. SAMPs, such as prostaglandin E2 (PGE2), annexin A1 (AnxA1), and specialized pro-resolving mediators (SPMs) inhibit the proliferation of proinflammatory cytokines, thereby aiding to manage inflammation and prevent further tissue damage instituted by DAMP signaling [36, 37]. These sequential signaling processes must be carefully coordinated and followed by DAMP-promoted tissue repair to avoid the development of disease [36]. If DAMP and SAMP signaling goes awry, sterile inflammation may become dysregulated. Prolonged DAMP and inadequate SAMP signaling may lead to hyperinflammation or chronic inflammation. On the other hand, immunosuppression from increased SAMP signaling may promote infection [36, 38]. An optimal “DAMP:SAMP ratio” must be achieved for wound healing without additional complications [36]. Land proposes balancing DAMP and SAMP levels to help heal inflammatory diseases, which may be accomplished by administering SAMPs [36, 39]. For now, therapeutic applications in the realm of cardiac transplantation remain unexplored.
Ischemia–reperfusion injury (IRI), a common consequence of transplantation
DAMPs are endogenous molecules that are released during stress, such as IRI, and act upon diverse innate immunity mediating receptors. Tissue injuries in HT relating to the initial state of the donor organ, preservation conditions, and potential ischemia–reperfusion injury (IRI) cause the release of various DAMPs [40]. The interaction between DAMPs and toll-like receptors (TLR) induces the release of pro-inflammatory cytokines, leading to the recruitment of neutrophils and macrophages at the site of injury to trigger an immune cascade [40, 41]. Sterile inflammation, or inflammation in the absence of a pathogen, contributes to DAMP release and subsequent allograft injury [13, 42, 43]
In IRI, ischemia during organ procurement results in cellular hypoxia and reduced ATP production. The lack of ATP disrupts the Na+–K+ ATPase pump and subsequently results in an influx of Na+ and water leading to cell swelling, dysregulated Ca2+, release, and disruption of cell membrane proteins [42]. Receptors in graft endothelial cells respond to the lack of blood flow in IRI by depolarizing the endothelial cell membrane, activation of NADPH oxidase, and production of reactive oxygen species (ROS) [44,45,46,47]. Reperfusion leads to the formation of superoxide radicals causing lipid peroxidation and the secretion of pro-inflammatory cytokines including CXCL1, CXCL2, TNF-α, IL-8, and IL-17 [48, 49]. The barrage of inflammatory cytokines worsens the inflammation already caused by ischemia.
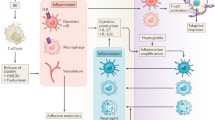
Following IRI in the heart, there is intramyocardial deposition of iron [42]. Any residual myocardial iron contributes to ferroptosis causing adverse cardiac remodeling post-reperfusion [42, 50, 51]. This process results in DAMP release and contributes to neutrophil adhesion to coronary vascular endothelial cells via the TLR4-Trif-Type I IFN-dependent signaling axis [42, 52, 53]. Li et al. have shown that treatment with ferrostatin-1, as well as coronary artery ligation, inhibited ferroptosis, improving systolic function and ventricular remodeling [42, 52]. Preventing ferroptosis will also reduce DAMP release that leads to further myocardial damage (Fig. 1).

IRI during cardiac transplantation results in DAMP release. Coordinated DAMP signaling cascades promote inflammation via activation of mediators. DAMPs, receptors, and mediators can be inactivated or blocked by experimental therapeutics
DAMPs in transplant rejection
DAMPs released from myocardial injuries, such as post-transplant IRI, lead to transplant rejection. Studies discovered histones, heat-shock proteins (HSPs), high-mobility group box 1 (HMGB1), DNA, rRNA, miRNA, and ATP as possible danger signals in transplantation aggravating the inflammation [40, 54]. HMGB1 is the most studied DAMP involved in transplant rejection. Following transplantation, HMGB1 is significantly upregulated [55]. HMGB1 is passively secreted from cells damaged in organ harvest and IRI and actively released by immune cells within the allograft. By interacting with DNA released by necrotic cells, HMGB1 upregulates pro-inflammatory pathways through PRRs such as TLR2, and TLR4 and induces macrophages-driven IL-23 [42, 45, 56, 57]. IL-23 stimulates γδ T cells to release IL-17A, which results in cardiomyocyte apoptosis and neutrophil recruitment [57]. Treatment with rA-box, an endogenous HMGB1 inhibitor, has been shown to delay heart allograft rejection by 1 week and reduced levels of proinflammatory cytokines including TNF-α [55]. Furthermore, inhibition of the HMGB1-IL-23/IL-17 pathway via glycyrrhizin or necrostatin-1 alleviates myocardial IRI suggesting the potential role of HMGB1 in IRI [42, 45, 58]. Necrostatin-1 is a non-specific necroptosis inhibitor that also inhibits ferroptosis [42]. HMGB1’s regulation of a variety of processes that culminate in IRI and graft rejection makes it an excellent therapeutic target to reduce potential myocardial damage.
ATP is the primary energy source used in most cellular functions. Small amounts of ATP exist extracellularly (eATP). The release of ATP from apoptotic or damaged endothelial cells and platelets after IRI disrupt the cell’s ability to carry out homeostasis [42]. eATP acts as a DAMP that binds to P2X7R and results in Nod-like receptor protein 3 (NLRP3) inflammasome activation and release of IL-1β during injury [42, 59]. Furthermore, eATP acts as a mitogen to induce cell cycle proliferation and tyrosine phosphorylation [11]. Vergani et al. evaluated the expression of P2X7R in cardiac transplants in murine models and noted its significant upregulation [60]. Next, short-term inhibition of P2X7R with periodate-oxidized ATP increased graft survival to greater than 100 days in 80% of murine transplant recipients [60, 61] (Table 2). Additionally, Vergani et al. noted the reduction of CD4+ T cells, CD8+ T cells, regulatory T cells, and Th17 [60]. Overall, the study has shown that the ATP/P2X7R pathway is another potential target that can improve cardiac transplant rejection and long-term graft survival [59, 60].
The S100 protein family is produced by cells of myeloid origin and has been linked to CAV. Several members of the family, such as S100A8, S100A9, S100A8/A9, S100A12, and S100A15, function as DAMPs that trigger endothelial cells and other cell types to produce inflammatory cytokines and chemokines [70]. Myeloid cell activation and tubulin-dependent translocation to the plasma membrane mediates S100 protein release [56]. Subsequently, S100 proteins lead to inflammation by binding to a variety of cell surface receptors such as RAGE and TLR4. Studies examining lung allograft stability have found the elevation of S100 proteins in patients with restrictive allograft syndrome compared to control patients with stable graft function [70]. In contrast, expression of S100A8 in kidney allografts was associated with a reduced incidence of CAV. Inhibition of RAGE with murine soluble RAGE (sRAGE) has been attributed to improvement of graft survival and a reduction in S100 [62, 71, 72]. Although the role of S100A8/A9 in HT has not been elucidated, it is potentially involved in atherogenesis, ischemia-associated MI, and heart failure [62, 70]. Due to its role in enhancing inflammation, S100A8/A9 potentially plays a role in heart transplant rejection (HTR); however, warrants further research [73].
Extracellular matrix molecules act as DAMPs in the setting of HTR. Molecules, such as heparan sulfate proteoglycan (HSPG) and fibronectin, enhance and modulate the pro-inflammatory state [54, 74]. HSPG is an acidic linear polysaccharide in the extracellular matrix and basement membrane of mammalian cells. HSPG and soluble heparan sulfate are heavily involved in tissue repair mechanisms and clearance of cellular debris. As DAMPs, these molecules interact with TLR4 to release proinflammatory cytokines and promote the maturation of dendritic cells [74, 75]. Fibronectin performs various functions, such as cell differentiation, proliferation, and extracellular matrix assembly [76]. When fibronectin functions as a DAMP, it recruits lymphocytes and helps induce their migration to the site of injury [77]. Chorawala et al. confirmed the importance of fibronectin as a proinflammatory signaling molecule using a mouse-model where 15 min post-transfusion anti-fibronectin antibodies significantly reduced IRI [63].
Hyaluronan, also known as hyaluronic acid (HA), is a ubiquitous extracellular matrix (ECM) glycosaminoglycan and acts as another DAMP that is released during injury and infection [14, 42]. HA degrades to high-weight (HMW-HA) and low-weight molecular forms (LMW-HA) [42, 45, 78]. The LMW-HA dominates in triggering pro-inflammatory events via TLR4 signaling to stimulate the maturation of monocyte-derived dendritic cells and upregulate TNF-α, IL-1β, and IL-12 [42]. Hällgren et al. visualized HA in rat heart grafts at various time points after transplantation. In normal, non-transplanted hearts, HA was seen in myocardial interstitial tissue and in the adventitia of arteries and veins [79]. By day 6 post-transplantation, cardiac allografts displayed threefold increase in HA levels in areas with edematous interstitial tissue due to leukocyte infiltration. Hackert et al. demonstrated the role of hyaluronan using 4-methylumbelliferone (4-MU), a naturally occurring coumarin that inhibits hyaluronan synthesis and deposition [69]. They observed that 4-MU reduces cardiac remodeling, perivascular and interstitial left ventricular fibrosis, and cardiomyocyte fibrosis. Although some studies have reported pro-inflammatory properties of HA, there are conflicting reports discussing its anti-inflammatory effects warranting further investigations [14, 78, 80, 81].
Cardiac transplant pathology and toll-like receptors
Studies have predominantly focused on the role of the adaptive immune response in AMR, though recently, investigations found a critical role of DAMP and TLR regulation activity in mediating the adaptive immune response in AMR [74]. Following the discovery of TLRs, the innate immune system has been at the forefront of immunological research [74, 82]. These receptors are expressed in a multitude of hematopoietic cells, endothelial cells and organ parenchymal cells [40, 41, 83]. Unlike T cell receptors and antibodies, TLRs lack a vast reserve of antigen recognition epitopes; however, they excel in immediate recognition of ligands and eliciting the downstream response.
Canonically, TLRs have been associated with antimicrobial defense against foreign pathogenic particles, such as lipopolysaccharide (LPS) or viral DNA and RNA. The most studied TLRs include TLR2 and TLR4. TLR2 forms heterodimers with TLR1 or TLR6 upon recognition of peptidoglycan on Gram-positive bacteria and bacterial lipoproteins [84]. TLR4 forms heterodimers with MD2 and CD14 upon recognition of LPS on Gram-negative bacteria. The function of TLRs has recently expanded to include interactions with DAMPs such as HMGB1 [85]. Studies of TLR4- and TLR2-deficient mice have found increased survival rates post-IRI compared to WT mice [42]. Although leukocyte infiltration was similar between deficient and WT groups, TLR2-deficient mice exhibited minimal myocardial damage in non-infarcted regions of the heart suggesting the regulatory roles of TLR2 and TLR4 in post-IRI myocardial damage and remodeling. Many similar studies have also identified potential therapeutics that target TLR2 and TLR4 signaling, thus attenuating myocardial damage from IRI (Table 3).
In transplantation, alloimmunity has been achieved by the knockout of MyD88, the universal TLR signal adapter protein [84]. By disarming MyD88 pro-inflammatory signaling pathways, such as those involving IL-1 and IL-18, Goldstein et al. demonstrated that TLRs are activated by molecules other than traditional PAMPs. Additionally, halting IL-18 signaling by MyD88 knockout could halt processes that mediate rejection, such as IRI [94]. Their study found that MyD88 knockout mice survived more than 100 days following skin transplant whereas wild-type mice rejected the allograft and survived a median of 16 days [84]. In addition, He et al. demonstrated similar results where a short course of MyD88 inhibitor, ST2825, significantly prolonged post-transplant survival of mouse cardiac allografts [95]. Moreover, the ST2825 treatment reduced necrosis and graft infiltration with inflammatory cytokines. Furthermore, Hsieh et al.’s mouse-model study on bone allograft rejection has described the upregulation of MyD88 and TLR-4 in femoral allografts [96]. Since the MyD88-TLR4 signaling pathway is upregulated in femoral allograft rejection, it may potentially be involved in the pathogenesis of cardiac transplant rejection. A seminal study by Frye et al. demonstrated that CCR2+ macrophages produce CXCL5 in a TLR9/MyD88-dependent fashion, promoting and regulating the extravasation of neutrophils [42, 97]. The upstream extravasation is mediated through TLR-4/Trif/type I IFN signaling pathways in endothelial cells that are activated through ferroptotic cell death of graft cells. Additionally, Li et al. have shown that neutrophil extravasation into inflamed myocardial tissue is also mediated via TLR9/MyD88/CXCL5 signaling [97]. Specifically, TLR9 regulates the expression of neutrophil chemoattractants, thereby contributing to the neutrophil trafficking responsible for IRI. MyD88 deficient macrophages can adequately inhibit neutrophil extravasation, preventing further damage to an already injured heart.
TLR downstream signaling effects include inflammasome assembly [42]. The inflammasome is a macromolecular, intracellular complex responsible for amplifying inflammatory signals and can be activated by TLR2, TLR3, TLR4, TNFR1, or IL-1R stimulation [42, 98]. Once priming of the inflammasome complex occurs through post translational modification of NLRP3 or Nuclear Factor-kB (NF-kB) augmentation of inflammasome components (NLRP3, caspase-1, and ASC) and cytokines (pro-IL-1b and pro-IL-18), the inflammasome produces caspase-1. Caspase-1 results in the production of IL-1b and IL-18, leading to inflammation [99]. Shah et al. performed biopsies of human hearts following cardiac transplant rejection which revealed inflammasome “specks” in 7 of 8 hearts [42, 98]. The presence of these “specks” indicates that inflammasome activation potentially has a role in cardiac transplant rejection.
DAMP binding to PRRs and TLRs activates the adaptive immune system through costimulatory molecules and cytokine stimulation of macrophages. Macrophages reprogrammed through epigenetic changes and metabolic signaling to develop immunologic memory and hypersensitivity to stimulation [42, 100]. Local modulation of macrophages through PRRs, dectin-1 and TLR4 has been shown to amplify this reprogramming, leading to transplant rejection. Dectin-1 has been localized to apoptotic tissues in rejection [42]. This is significant since apoptotic tissues in donor organs upregulate the production of vimentin, an endogenous molecule involved in wound healing which is a potent ligand for dectin-1 [42, 100, 101]. In addition, HMGB1 signaling contributes to the reprogramming of immunity leading to the release of proinflammatory cytokines [42].
Conclusion and future directions
Rejection is a major complication following solid-organ transplant. Review of current literature on the role of the innate immune system in cardiac allograft rejection indicates the potency of DAMPs and their corresponding TLRs in the initial alloimmune response associated with HTR. DAMPs play a significant role in the initial stages of solid-organ transplant rejection by eliciting a rapid immune response. Currently, most studies explore the role of DAMPs in kidney and lung transplantation. Although DAMP pathways in cardiac transplant rejection remain unexplored, their implications in IRI and other transplanted organ rejections suggest that the transplanted heart most likely responds in a similar manner.
Considering the lack of therapies targeting the innate immune system, a better understanding of DAMP signaling pathways in HT failure may support potential interventions in the future transplantation medicine. Therapeutics that block the initial DAMP release may prevent or delay further complications along the signaling pathway. Furthermore, blocking DAMP-TLR interactions possibly prevent the downstream inflammatory effects of this interaction. Investigation on DAMPs, and their corresponding TLRs, would open beneficial therapeutic regimes to patients who are resistant to the current available options. Drugs that possibly target DAMP binding and signal transduction could prevent the devastating effects of acute allograft rejection. Encouragingly, emerging research in the involvement of DAMPs in HTR could open several translational avenues in the management of transplant failure.
Data availability
Not applicable; all information is gathered from published articles.
Code availability
Not applicable.
References
World Health Organization. Cardiovascular diseases (CVDs). Fact sheet. https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed 11 June 2021
Virani SS, Alonso A, Aparicio HJ et al (2021) Heart disease and stroke statistics—2021 update: a report from the American Heart Association. Circulation. https://doi.org/10.1161/CIR.0000000000000950
Heidenreich PA, Trogdon JG, Khavjou OA et al (2011) Forecasting the future of cardiovascular disease in the United States. Circulation 123:933–944. https://doi.org/10.1161/CIR.0b013e31820a55f5
Taylor CJ, Ordóñez-Mena JM, Roalfe AK et al (2019) Trends in survival after a diagnosis of heart failure in the United Kingdom 2000–2017: population based cohort study. BMJ 364:223. https://doi.org/10.1136/bmj.l223
Alraies MC, Eckman P (2014) Adult heart transplant: indications and outcomes. J Thorac Dis 6:1120–1128. https://doi.org/10.3978/j.issn.2072-1439.2014.06.44
Li J, Li C, Zhuang Q et al (2019) The evolving roles of macrophages in organ transplantation. J Immunol Res 2019:1–11
Fuchs M, Schibilsky D, Zeh W et al (2019) Does the heart transplant have a future? Eur J Cardio-Thorac Surg 55:i38–i48. https://doi.org/10.1093/ejcts/ezz107
Colvin MM, Cook JL, Chang P et al (2015) Antibody-mediated rejection in cardiac transplantation: emerging knowledge in diagnosis and management. Circulation. https://doi.org/10.1161/CIR.0000000000000093
Garces JC, Giusti S, Staffeld-Coit C et al (2017) Antibody-mediated rejection: a review. Ochsner J 17:46–55
Schinstock CA, Mannon RB, Budde K et al (2020) Recommended treatment for antibody-mediated rejection after kidney transplantation: the 2019 expert consensus from the Transplantion Society Working Group. Transplantation 104:911–922. https://doi.org/10.1097/TP.0000000000003095
Patel S (2018) Danger-associated molecular patterns (DAMPs): the derivatives and triggers of inflammation. Curr Allergy Asthma Rep 18:63. https://doi.org/10.1007/s11882-018-0817-3
Todd JL, Palmer SM (2017) Danger signals in regulating the immune response to solid organ transplantation. J Clin Investig 127:2464–2472. https://doi.org/10.1172/JCI90594
Zindel J, Kubes P (2020) DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 15:493–518. https://doi.org/10.1146/annurev-pathmechdis-012419-032847
Dwyer GK, Turnquist HR (2021) Untangling local pro-inflammatory, reparative, and regulatory damage-associated molecular-patterns (DAMPs) pathways to improve transplant outcomes. Front Immunol 12:49. https://doi.org/10.3389/fimmu.2021.611910
Tsilimingas NB (2003) Modification of bicaval anastomosis: an alternative technique for orthotopic cardiac transplantation. Ann Thorac Surg 75:1333–1334. https://doi.org/10.1016/S0003-4975(02)04550-2
Calderwood SK, Gong J, Murshid A (2016) Extracellular HSPs: the complicated roles of extracellular HSPs in immunity. Front Immunol 7:159. https://doi.org/10.3389/fimmu.2016.00159
Calderwood SK, Murshid A, Gong J (2012) Heat shock proteins: conditional mediators of inflammation in tumor immunity. Front Immunol. https://doi.org/10.3389/fimmu.2012.00075
Millington TM, Madsen JC (2009) Innate immunity in heart transplantation. Curr Opin Organ Transplant 14:571–576. https://doi.org/10.1097/MOT.0b013e32832e7158
Murao A, Aziz M, Wang H et al (2021) Release mechanisms of major DAMPs. Apoptosis 26:152–162. https://doi.org/10.1007/s10495-021-01663-3
Yuan S, Liu Z, Xu Z et al (2020) High mobility group box 1 (HMGB1): a pivotal regulator of hematopoietic malignancies. J Hematol Oncol 13:91. https://doi.org/10.1186/s13045-020-00920-3
Parisi L, Toffoli A, Ghezzi B et al (2020) A glance on the role of fibronectin in controlling cell response at biomaterial interface. Jpn Dent Sci Rev 56:50–55. https://doi.org/10.1016/j.jdsr.2019.11.002
Gupta RC, Lall R, Srivastava A, Sinha A (2019) Hyaluronic acid: molecular mechanisms and therapeutic trajectory. Front Vet Sci 6:192. https://doi.org/10.3389/fvets.2019.00192
Simon Davis DA, Parish CR (2013) Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol. https://doi.org/10.3389/fimmu.2013.00470
Jang HS, Shin WJ, Lee JE, Do JT (2017) CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8:148. https://doi.org/10.3390/genes8060148
Kim I-C, Youn J-C, Kobashigawa JA (2018) The past, present and future of heart transplantation. Korean Circ J 48:565–590. https://doi.org/10.4070/kcj.2018.0189
Sarris GE, Moore KA, Schroeder JS et al (1994) Cardiac transplantation: the Stanford experience in the cyclosporine era. J Thorac Cardiovasc Surg 108:240–252. https://doi.org/10.1016/S0022-5223(94)70006-0
Söderlund C, Rådegran G (2015) Immunosuppressive therapies after heart transplantation—the balance between under- and over-immunosuppression. Transplant Rev 29:181–189. https://doi.org/10.1016/j.trre.2015.02.005
Tonsho M, Michel S, Ahmed Z et al (2014) Heart transplantation: challenges facing the field. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a015636
Westerdahl DE, Kobashigawa JA (2019) Heart transplantation for advanced heart failure. Cardiac Intensive Care. https://doi.org/10.1016/B978-0-323-52993-8.00048-5
Gupta T, Krim SR (2019) Cardiac transplantation: update on a road less traveled. Ochsner J 19:369–377. https://doi.org/10.31486/toj.19.0022
Bresnick AR (2018) S100 proteins as therapeutic targets. Biophys Rev 10:1617–1629. https://doi.org/10.1007/s12551-018-0471-y
Rosenberg JH, Rai V, Dilisio MF, Agrawal DK (2017) Damage associated molecular patterns in the pathogenesis of osteoarthritis: potentially novel therapeutic targets. Mol Cell Biochem 434:171–179. https://doi.org/10.1007/s11010-017-3047-4
Andersson U, Yang H, Harris H (2018) Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets 22:263–277. https://doi.org/10.1080/14728222.2018.1439924
Hayashi K, Nikolos F, Lee YC et al (2020) Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat Commun 11:6299. https://doi.org/10.1038/s41467-020-19970-9
Yin P, Wang X, Wang S et al (2019) Maresin 1 improves cognitive decline and ameliorates inflammation in a mouse model of Alzheimer’s disease. Front Cell Neurosci 13:466. https://doi.org/10.3389/fncel.2019.00466
Land WG (2020) Use of DAMPs and SAMPs as therapeutic targets or therapeutics: a note of caution. Mol Diagn Ther 24:251–262. https://doi.org/10.1007/s40291-020-00460-z
Varki A (2011) Letter to the Glyco-Forum: since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology 21:1121–1124. https://doi.org/10.1093/glycob/cwr087
Coady A, Nizet V (2016) SAMP-ening down sepsis. Ann Transl Med 4:31–31. https://doi.org/10.21037/atm.2016.11.31
Relja B, Land WG (2020) Damage-associated molecular patterns in trauma. Eur J Trauma Emerg Surg 46:751–775. https://doi.org/10.1007/s00068-019-01235-w
Land WG, Agostinis P, Gasser S et al (2016) Transplantation and damage-associated molecular patterns (DAMPs). Am J Transplant 16:3338–3361. https://doi.org/10.1111/ajt.13963
Sosa RA, Rossetti M, Naini BV et al (2018) Pattern recognition receptor–reactivity screening of liver transplant patients: potential for personalized and precise organ matching to reduce risks of ischemia–reperfusion injury. Ann Surg. https://doi.org/10.1097/SLA.0000000000003085.10.1097/SLA.0000000000003085
Frye CC, Bery AI, Kreisel D, Kulkarni HS (2021) Sterile inflammation in thoracic transplantation. Cell Mol Life Sci 78:581–601. https://doi.org/10.1007/s00018-020-03615-7
Chen GY, Nuñez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837. https://doi.org/10.1038/nri2873
Bhogal RH, Weston CJ, Velduis S et al (2018) The reactive oxygen species-mitophagy signaling pathway regulates liver endothelial cell survival during ischemia/reperfusion injury. Liver Transpl 24:1437–1452. https://doi.org/10.1002/lt.25313
Braza F, Brouard S, Chadban S, Goldstein DR (2016) Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat Rev Nephrol. https://doi.org/10.1038/nrneph.2016.41
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia–reperfusion injury: a neglected therapeutic target. J Clin Investig 123:92–100. https://doi.org/10.1172/JCI62874
Granger DN, Kvietys PR (2015) Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 6:524–551. https://doi.org/10.1016/j.redox.2015.08.020
Sosa RA, Zarrinpar A, Rossetti M et al (2016) Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight 1(20):e89679. https://doi.org/10.1172/jci.insight.89679
Chen W, Li D (2020) Reactive oxygen species (ROS)-responsive nanomedicine for solving ischemia–reperfusion injury. Front Chem 8:732. https://doi.org/10.3389/fchem.2020.00732
Bulluck H, Rosmini S, Abdel-Gadir A et al (2016) Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment–elevation myocardial infarction and adverse left ventricular remodeling. Circulation. https://doi.org/10.1161/CIRCIMAGING.116.004940
Yan H-F, Tuo Q-Z, Yin Q-Z, Lei P (2020) The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool Res 41:220–230. https://doi.org/10.24272/j.issn.2095-8137.2020.042
Li W, Feng G, Gauthier JM et al (2019) Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Investig 129(6):2293–2304. https://doi.org/10.1172/JCI126428
Tang D, Kepp O, Kroemer G (2021) Ferroptosis becomes immunogenic: implications for anticancer treatments. Oncoimmunology. https://doi.org/10.1080/2162402X.2020.1862949
Schaefer L (2014) Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem 289:35237–35245. https://doi.org/10.1074/jbc.R114.619304
Huang Y, Yin H, Han J et al (2007) Extracellular Hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant 7:799–808. https://doi.org/10.1111/j.1600-6143.2007.01734.x
Bertheloot D, Latz E (2017) HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol 14:43–64. https://doi.org/10.1038/cmi.2016.34
Zhu H, Li J, Wang S, Liu K et al (2013) Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia–reperfusion injury in a cardiac transplantation model. Transplantation 95:1448–1454. https://doi.org/10.1097/TP.0b013e318293b7e1
Zhang A, Mao X, Li L et al (2014) Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl Int 27:1077–1085. https://doi.org/10.1111/tri.12349
Vergani A, Tezza S, Fotino C et al (2014) The purinergic system in allotransplantation. Am J Transplant 14:507–514. https://doi.org/10.1111/ajt.12567
Vergani A, Tezza S, D’Addio F et al (2013) Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation 127:463–475. https://doi.org/10.1161/CIRCULATIONAHA.112.123653
Zeiser R, Robson SC, Vaikunthanathan T et al (2016) Unlocking the potential of purinergic signaling in transplantation. Am J Transplant 16:2781–2794. https://doi.org/10.1111/ajt.13801
Silvis MJM, Kaffka Genaamd Dengler SE, Odille CA et al (2020) Damage-associated molecular patterns in myocardial infarction and heart transplantation: the road to translational success. Front Immunol 11:599511. https://doi.org/10.3389/fimmu.2020.599511
Chorawala MR, Prakash P, Doddapattar P et al (2018) Deletion of extra domain A of fibronectin reduces acute myocardial ischemia/reperfusion injury in hyperlipidemic mice by limiting thrombo-inflammation. Thromb Haemost 118:1450–1460. https://doi.org/10.1055/s-0038-1661353
Qin C-Y, Zhang H-W, Gu J et al (2017) Mitochondrial DNA-induced inflammatory damage contributes to myocardial ischemia reperfusion injury in rats: cardioprotective role of epigallocatechin. Mol Med Rep 16:7569–7576. https://doi.org/10.3892/mmr.2017.7515
Chen C, Feng Y, Zou L et al (2014) Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia–reperfusion injury. J Am Heart Assoc 3:e000683. https://doi.org/10.1161/JAHA.113.000683
Chen HH, Yuan H, Cho H et al (2017) Theranostic nucleic acid binding nanoprobe exerts anti-inflammatory and cytoprotective effects in ischemic injury. Theranostics 7:814–825. https://doi.org/10.7150/thno.17366
Tu G, Zou L, Liu S et al (2016) Long noncoding NONRATT021972 siRNA normalized abnormal sympathetic activity mediated by the upregulation of P2X7 receptor in superior cervical ganglia after myocardial ischemia. Purinergic Signal 12:521–535. https://doi.org/10.1007/s11302-016-9518-3
Gu M, Zheng A-B, Jin J et al (2016) Cardioprotective effects of genistin in rat myocardial ischemia–reperfusion injury studies by regulation of P2X7/NF-κB pathway. Evid Based Complement Altern Med 2016:5381290. https://doi.org/10.1155/2016/5381290
Hackert K, Homann S, Mir S et al (2021) 4-Methylumbelliferone attenuates macrophage invasion and myocardial remodeling in pressure-overloaded mouse hearts. Hypertension 77:1918–1927. https://doi.org/10.1161/HYPERTENSIONAHA.120.15247
Schiopu A, Cotoi OS (2013) S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Med Inflamm 2013:1–10
Moser B, Szabolcs MJ, Ankersmit HJ et al (2007) Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am J Transplant 7:293–302. https://doi.org/10.1111/j.1600-6143.2006.01617.x
Volz HC, Laohachewin D, Seidel C et al (2012) S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-κB signaling. Basic Res Cardiol 107:250. https://doi.org/10.1007/s00395-012-0250-z
Wang S, Song R, Wang Z et al (2018) S100A8/A9 in inflammation. Front Immunol. https://doi.org/10.3389/fimmu.2018.01298
Brennan TV, Lunsford KE, Kuo PC (2010) Innate pathways of immune activation in transplantation. J Transplant. https://doi.org/10.1155/2010/826240
Matta BM, Reichenbach DK, Blazar BR, Turnquist HR (2017) Alarmins and their receptors as modulators and indicators of alloimmune responses. Am J Transplant 17:320. https://doi.org/10.1111/ajt.13887
Pankov R, Yamada KM (2002) Fibronectin at a glance. J Cell Sci 115:3861–3863. https://doi.org/10.1242/jcs.00059
Kostrzewa-Nowak D, Ciechanowicz A, Clark JSC, Nowak R (2020) Damage-associated molecular patterns and Th-cell-related cytokines released after progressive effort. J Clin Med 9:876. https://doi.org/10.3390/jcm9030876
Collins SL, Black KE, Chan-Li Y et al (2011) Hyaluronan fragments promote inflammation by down-regulating the anti-inflammatory A2a receptor. Am J Respir Cell Mol Biol 45:675–683. https://doi.org/10.1165/rcmb.2010-0387OC
Hällgren R, Gerdin B, Tengblad A, Tufveson G (1990) Accumulation of hyaluronan (hyaluronic acid) in myocardial interstitial tissue parallels development of transplantation edema in heart allografts in rats. J Clin Investig 85:668–673. https://doi.org/10.1172/JCI114490
Petrey A, de la Motte C (2014) Hyaluronan, a crucial regulator of inflammation. Front Immunol 5:101. https://doi.org/10.3389/fimmu.2014.00101
Ruppert SM, Hawn TR, Arrigoni A et al (2014) Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol Res 58:186–192. https://doi.org/10.1007/s12026-014-8495-2
Liu X, Cao H, Li J et al (2017) Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia–reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. https://doi.org/10.1038/cdd.2017.1
LaRosa DF, Rahman AH, Turka LA (2007) The innate immune system in allograft rejection and tolerance. J Immunol. https://doi.org/10.4049/jimmunol.178.12.7503
Goldstein DR, Tesar BM, Akira S, Lakkis FG (2003) Critical role of the toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Investig. https://doi.org/10.1172/JCI17573
Georgel P (2016) Innate immune receptors in solid organ transplantation. Hum Immunol 77:1071–1075. https://doi.org/10.1016/j.humimm.2016.04.004
Arslan F, Smeets MB, O’Neill LAJ et al (2010) Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 121:80–90. https://doi.org/10.1161/CIRCULATIONAHA.109.880187
Hennessy EJ, Parker AE, O’Neill LAJ (2010) Targeting toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 9:293–307. https://doi.org/10.1038/nrd3203
Mistry P, Laird MHW, Schwarz RS et al (2015) Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci USA 112:5455–5460. https://doi.org/10.1073/pnas.1422576112
Gao H-K, Yin Z, Zhang R-Q et al (2009) GSK-3β inhibitor modulates TLR2/NF-κB signaling following myocardial ischemia–reperfusion. Inflamm Res 58:377–383. https://doi.org/10.1007/s00011-009-0002-1
Shimamoto A, Chong AJ, Yada M et al (2006) Inhibition of toll-like receptor 4 with eritoran attenuates myocardial ischemia–reperfusion injury. Circulation 114:1–270. https://doi.org/10.1161/CIRCULATIONAHA.105.000901
Li C, Ha T, Kelley J et al (2004) Modulating toll-like receptor mediated signaling by (1→3)-β-d-glucan rapidly induces cardioprotection. Cardiovasc Res 61:538–547. https://doi.org/10.1016/j.cardiores.2003.09.007
Bai Y, Li Z, Liu W et al (2019) Biochanin a attenuates myocardial ischemia/reperfusion injury through the TLR4/NF-κB/NLRP3 signaling pathway. Acta Cir Bras. https://doi.org/10.1590/s0102-865020190110000004
Wang Y-H, Chen K-M, Chiu P-S et al (2016) Lumbrokinase attenuates myocardial ischemia–reperfusion injury by inhibiting TLR4 signaling. J Mol Cell Cardiol 99:113–122. https://doi.org/10.1016/j.yjmcc.2016.08.004
Liu C, Chen J, Liu B et al (2018) Role of IL-18 in transplant biology. Eur Cytokine Netw 29:48–51. https://doi.org/10.1684/ecn.2018.0410
He W-T, Zhang L-M, Li C et al (2016) Short-term MyD88 inhibition ameliorates cardiac graft rejection and promotes donor-specific hyporesponsiveness of skin grafts in mice. Transpl Int 29:941–952. https://doi.org/10.1111/tri.12789
Hsieh J-L, Shen P-C, Wu P-T et al (2017) Knockdown of toll-like receptor 4 signaling pathways ameliorate bone graft rejection in a mouse model of allograft transplantation. Sci Rep 7:46050. https://doi.org/10.1038/srep46050
Li W, Hsiao H-M, Higashikubo R et al (2016) CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1(12):e87315. https://doi.org/10.1172/jci.insight.87315
Shah KB, Mauro AG, Flattery M et al (2015) Formation of the inflammasome during cardiac allograft rejection. Int J Cardiol 201:328–330. https://doi.org/10.1016/j.ijcard.2015.08.070
Cheung KP, Kasimsetty SG, McKay DB (2013) Innate immunity in donor procurement. Curr Opin Organ Transplant 18:154–160. https://doi.org/10.1097/MOT.0b013e32835e2b0d
Ochando J, Fayad ZA, Madsen JC et al (2020) Trained immunity in organ transplantation. Am J Transplant 20:10–18. https://doi.org/10.1111/ajt.15620
Rose ML (2004) De novo production of antibodies after heart or lung transplantation should be regarded as an early warning system. J Heart Lung Transplant 23:385–395. https://doi.org/10.1016/j.healun.2003.08.028
Acknowledgements
The figure was created using BioRender.com
Funding
The research work of DK Agrawal is supported by NIH-NHLBI Grants R01HL147662 and R01HL144125. The content of this original review article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
Conception and design: FGT; Literature search, collection of the scientific information, analysis, and interpretation of the data: AK, DKA, FGT; Drafting of the article: AK, FGT; Critical revision and editing of the article for important intellectual content: FGT, DKA; Final approval of the submitted article: AK, DKA, FGT.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kesler, A., Agrawal, D.K. & Thankam, F.G. Toll-like receptors and damage-associated molecular patterns in the pathogenesis of heart transplant rejection. Mol Cell Biochem 477, 2841–2850 (2022). https://doi.org/10.1007/s11010-022-04491-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04491-4