
Abstract
Objective
The present study defines the expression of Toll-like Receptor 2 (TLR2), and the modulatory role of Glycogen synthase kinase (GSK)-3β inhibitor on TLR2/Nuclear Factor-kappa B (NF-κB) signaling following myocardial ischemia-reperfusion (MI-R) injury in rats.
Methods
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) and immunohistochemistry (IHC) were used to analyze the presence and quantity of TLR2 mRNA and protein. Tumor necrosis factor (TNF)-α mRNA and interleukin-6 (IL-6) mRNA were analyzed by RT-PCR. The activation of NF-κB was detected by Western Blot and the myocardial infarct size by Evans blue-TTC staining.
Results
Following 30 min of myocardial ischemia, a significant up-regulation of TLR2 mRNA was revealed by RT-PCR from 1 to 24 h post reperfusion. IHC demonstrated high protein expression levels of TLR2. Administration of the GSK-3β inhibitor 4-benzyl-2-methyl-1, 2, 4-thiadiazolidine-3, 5-dione (TDZD-8) 5 min prior to reperfusion following 1 h reperfusion down-regulated mRNA levels of TLR2 and downstream proinflammatory cytokines (P < 0.05 vs. MI-R), decreased the activity of NF-κB and the size of the myocardial infarct (P < 0.05 vs. MI-R).
Conclusion
Our results demonstrate that TLR2 and its signaling components are activated by MI-R. TDZD-8 administration attenuates TLR2/NF-κB signaling, suggesting a possible mechanism whereby GSK-3β inhibition improves the outcome of MI-R.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronary heart disease (CHD), the number one killer in the world, accounts for ~7.22 million deaths. The primary pathological manifestation of CHD is myocardial damage, most likely due to myocardial ischemia-reperfusion (MI-R) injury. For several decades it is known that the inflammatory response during reperfusion is the major cause of MI-R injury. Recently, the acute phase of I-R has been viewed as part of the innate immune response, with a lack of vascular perfusion and oxygenation. These non-infectious stimuli contribute to the inflammatory response, in part by signaling via Toll-like receptors (TLRs)-mediated pathways [1]. The identification of TLRs on cardiomyocytes not only has brought new insights on the inflammatory response initiated by cardiomyocytes themselves, but also offered potential targets to reduce I-R injury.
Toll-like receptors are widely conserved in organisms (ranging from sponges to humans) and are expressed on innate and adaptive immune system cells as well as on parenchymal cells, positioning them as molecular signals of invading microorganisms through pathogen-associated molecular patterns (PAMPs) and reacting to tissue damage signals (damage-associated molecular patterns; DAMPs). Damaged (ischemic) tissue is thought to release DAMPs, which activate TLRs, leading to downstream activation of NF-κB. This activation of NF-κB is known to play a vital role in innate immunity and participate in the inflammatory cascade [2]. Several molecules have been implicated as DAMPs, including heat shock proteins and necrotic cells. Interestingly, endogenous cellular injury signals, which can activate TLR2 and TLR4, are known to accumulate upon cardiac ischemic injury. Several reports have suggested that TLR2 activation is an important modulator in ischemic models [3, 4]. In particular, the absence of TLR2 has been shown to decrease cardiac ischemia [5, 6], and dysfunction related to doxorubicin toxicity [7]. These studies indicate that expression of TLR2 is intimately linked to immune responses of the heart.
Glycogen synthase kinase-3 (GSK-3) is a ubiquitously expressed serine/threonine kinase originally identified as a regulator of glycogen synthesis [8]. Administration of GSK-3β inhibitors, to mimic the effect of Ser9 phosphorylation of GSK-3β, has been shown to reduce necrotic cell production in the heart. Recently GSK-3β has emerged as a key regulatory switch in the modulation of the inflammatory response. Martin et al. [9] recently reported that GSK-3β inhibition reduced the production of proinflammatory cytokines and increased the anti-inflammatory cytokine IL-10 in human peripheral blood mononuclear cells stimulated with TLR2, TLR4, TLR5, or TLR9 agonists. Till date, there are no other studies on the role of TLR2 in MI-R injury documented. These data led us to investigate the hypothesis whether GSK-3β inhibitor administration could modulate TLR2/NF-κB signaling following MI-R injury in rats.
Materials and methods
All procedures were performed in accordance with the National Institute of Health Guideline on the Use of Laboratory Animals and were approved by the Fourth Military Medical University Committee on Animal Care.
Rat MI-R injury model
Male Sprague-Dawley rats weighing 200–250 g were obtained from the Experimental Animal Center of the Fourth Military Medical University and were divided into two groups, sham (n = 20) and MI-R (n = 30). In the MI-R group, hearts were exposed to a 30 min period of ischemia followed by reperfusion for 1, 6, 12, or 24 h. To produce I-R, rats were anesthetized with pentobarbital sodium (40 mg/kg, ip), Electrocardiograms (ECG) and heart rate (HR) were simultaneously recorded on a polygraph. Surgical preparation of myocardial ischemia was performed similarly to what was previously reported [10]. In brief, the chest was opened through a left thoracotomy, and the heart was exposed through a pericardiectomy. The left anterior descending coronary artery (LAD) was ligated with a 6-0 silk suture by passing a needle underneath the LAD and 2–3 mm inferior to the left auricle. The suture was removed after 30 min to allow for reperfusion of the heart muscle. In the sham group, the needle was passed under the LAD, however, occlusion was not performed. Significant ECG alterations, accompanied by color changes of the area at risk were considered a successful coronary occlusion. At the above time periods of reperfusion, the rats were re-anesthetized and sacrificed. Ischemic left ventricular tissue was used for real-time RT-PCR of TLR2mRNA and immunohistochemical staining for the protein expression of TLR2.
In protocol 2, rats were randomized to three groups (n = 20 each): (1) Sham + vehicle: 10% dimethyl sulfoxide (DMSO, 1 ml/kg, i.v.) and subjected to the surgical procedure; (2) I-R + vehicle: 30 min of ischemia followed by 1 h reperfusion with DMSO (1 ml/kg, i.v.); (3) TDZD-8: 30 min of ischemia followed by 1 h reperfusion with TDZD-8 (1 mg/kg, i.v., dissolved in DMSO, obtained from Calbiochem, Merck Biosciences Ltd, Beeston, Nottingham, UK) for 5 min prior to reperfusion. The doses of TDZD-8 used to reduce I-R injury were based on previous in vivo studies [11]. At the end of reperfusion, we observed the effect of TDZD-8 on mRNA levels of TLR2, TNF-α, and IL-6 by real-time RT-PCR and on the activation of NF-κB by Western Blot. Myocardial infarct size was examined by Evans blue-TTC staining.
Real time-reverse transcription polymerase chain reaction (RT-PCR)
Real-time PCR was performed in an ABI PRISM 7500 Sequence Detection System Thermal Cycler (Applied Biosystems). Total RNA was isolated from heart tissue and one microgram of total RNA was reverse transcribed by a RT-PCR kit according to the manufacture’s instructions (Promega, USA). Real-time PCR was performed using the SYBR Green fluorescence dye method, with the primers listed in Table 1. Two microliters of each cDNA template were amplified in a 25 µl reaction volume. SYBR green fluorescence was monitored after each elongation period, with the housekeeping gene found to be constantly expressed in the heart. Samples were amplified with β-actin primers for determination of the initial relative quantity of cDNA in each sample, and all PCR products were normalized to that amount. Fold increases in the expression of specific mRNAs in the MI-R heart compared to the sham hearts were calculated as 2−(ΔΔCT). Data are expressed as RQ (relative quantity) and differences are shown in the figures as the expression ratio of the normalized target gene.
Western Blot analyses
Whole cell tissue homogenates were obtained from snap-frozen LV tissue and separated using SDS-PAGE in phosphate-buffered saline. Samples were transferred to PVDF (polyvinylidene difluoride)-plus membranes, incubated with 5% milk Tris-buffered saline with Tween (TBST) for 1 h and membranes were incubated overnight with monoclonal antibodies against phospho-NF-κB p65 (ser 536, Cell Signaling, Danvers, MA, 1:1000) overnight at 4°C in TBST. After washing three times in TBST, membranes were incubated with an anti-rabbit-horseradish peroxidase-conjugated secondary antibody for 1 h at 37°C. Immune complexes were visualized with an enhanced chemiluminescence reagent. Results were quantified by capturing the exposed X-ray film image and optical densities were obtained using area measurements obtained from Image Pro Plus software.
Immunohistochemical staining
Tissue samples were fixed with 10% neutral buffered formalin and embedded in paraffin. The tissue sections (4 μm) were blocked with endogenous peroxidase using 3% hydrogen peroxide for 20 min. Sections were then washed three times in PBS and briefly in a buffer containing 1% polymerized bovine albumin and incubated with an anti-TLR2 polyclonal antibody (diluted 1:100, Santa Cruz Biotechnology) at 37°C for 2 h. After washing with PBS, each of the sections was incubated with horseradish peroxidase (HRP)-conjugated anti-goat IgG for 60 min at room temperature. Diaminobenzidine (DAB) was used as the chromogen and counterstaining was performed with haematoxylin. Six fields were randomly selected for each section and observed using a light microscope (×200).
Evaluation of myocardial infarct size
At the end of 1 h reperfusion, the ligature around the LAD was retightened and 1 ml of 2% Evans blue dye was injected into the side arm of the aortic cannula. The dye was present throughout the circulation, except in the region occluded (area-at-risk, AAR). The heart was then frozen at –20°C and sliced into 1 mm thick sections. These slices were incubated in 1% triphenyltetrazolium chloride (TTC) at 37°C for 30 min and photographed with a digital camera. Evans blue stained areas (area-not-at-risk, ANAR), TTC-stained areas (red staining, ischemic but viable tissue), and TTC negative areas (infarcted myocardium) in each slice were measured digitally using Image Pro Plus software (Media Cybernetics). AAR was expressed as a percentage of the left ventricular area (AAR/LV), and infarct size (IS) was expressed as a percentage of total AAR (IS/AAR).
Statistical analyses
All data are presented as mean ± SEM. Differences were compared by ANOVA or Student’s t-test where appropriate. P < 0.05 was considered to be statistically significant. All of the statistical tests were performed using GraphPad Prism software version 4.0 (GraphPad Software, San Diego, CA).
Results
MI-R increased TLR2 expression
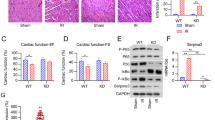
TLR2 was constitutively expressed in the sham group (Fig. 1a). After reperfusion, TLR2 mRNA expression was greater and increased over time (Fig. 1a). At 1, 6, 12, and 24 h reperfusion, expression of the TLR2 gene was 2.1-, 2.9-, 4.7- and 3.5-fold higher compared to the sham group (P < 0.001), respectively. Interestingly, immunohistochemical staining confirmed the expression of TLR2 occurred mainly in cardiac myocytes (Fig. 1b). Control myocytes exhibited diffuse staining with an anti-TLR2 polyclonal antibody, and the stainning was inhibited when a control antiserum was used (Fig. 1b). Upon immunohistochemical analysis, the diffuse pattern of TLR2 expression in the control tissue was noted to significantly change after 12 h reperfusion. In cardiac sections adjacent to the site of ischemic injury, enhanced and predominantly sarcolemmal staining of TLR2 was observed (Fig. 1b). Scattered foci of intense TLR2 staining involving 2 or more contiguous myocytes were noted in the I-R tissue, a feature not observed in sections from hearts of control animals.

a RT-PCR shows the relative expression of TLR2 mRNA. The expression of TLR2 in shams is regarded as the standard control (RQ = 1). TLR2 in hearts after reperfusion is expressed as a multiple of shams. b IHC analysis of TLR2 in cardiac muscle of rats. Photomicrographs are shown of rat cardiac muscle (×200), stained with a polyclonal anti-TLR2 antibody. Substitution of the primary antibody with a control cow antiserum (control serum) was used as a control. Positively stained cells were visualized with DAB (brown). Bars represent mean ± SEM ≥ 5 animals in each group. * P < 0.001 compared with sham
TDZD-8 decreases TLR2 mRNA and downstream pro-inflammatory cytokines
TLR2 is linked to NF-κB activation in several cell types and is correlated with expression of multiple pro-inflammatory cytokines, known to play important roles in myocardial remodeling. To investigate whether the functional effects of TLR2 are associated with pro-inflammatory cytokine production in MI-R, the levels of tissue TNF-α and IL-6 mRNA were measured after 30 min ischemia and 1 h reperfusion by RT-PCR. As shown in Fig. 2a–c, these mRNA levels were significantly increased after 1 h reperfusion (TLR2: 2.1 ± 0.1-fold higher than sham; TNF-α: 4.5 ± 0.4-fold higher than sham, and IL-6: 4.3 ± 0.4-fold higher than sham, P < 0.05). The mRNA levels of TNF-α and IL-6 were positively correlated with TLR2 mRNA levels after 1 h reperfusion (Fig. 2d, e). (TLR2 vs. TNF-α mRNA: r = 0.65, P < 0.05; TLR2 vs. IL-6 mRNA: r = 0.68, P < 0.05). Treatment with TDZD-8 had a significant effect on the mRNA levels of TLR2, TNF-α and IL-6 compared with MI-R (2.1 ± 0.1 to 1.5 ± 0.1; 4.5 ± 0.4 to 3.0 ± 0.5; 4.3 ± 0.4 to 2.7 ± 0.3; respectively, P < 0.05) (Fig. 2a–c).

Correlation plot of TLR2 mRNA with TNF-α and IL-6 mRNA and the effect of TDZD-8 on mRNA levels of TLR2 and downstream pro-inflammatory cytokines in rats after MI-R. Results are expressed as mean ± SEM n ≥ 6 animals from each group. * P < 0.05 compared with shams; # P < 0.05 compared with MI-R animals
TDZD-8 decreases NF-κB activity
All TLRs activate a common signaling pathway that ultimately activates NF-κB transcription factors. As the most important downstream molecule in the TLRs signaling pathway, NF-κB is a transcriptional factor required for the gene expression of many inflammatory mediators. Therefore, we were interested in the activity of NF-κB in our study. Studies have shown that p65 phosphorylation promotes NF-κB activation upon stimuli. The levels of phospho-NF-κB p65 protein in the nuclear fractions of the myocardial tissue were substantially elevated by MI-R compared to the sham group (P < 0.05), as determined by Western Blot (Fig. 3a). Furthermore, there was a significant correlation between TLR2 mRNA levels and the levels of phospho-NF-κB p65 protein after 1 h reperfusion (Fig. 3b). (TLR2 mRNA vs. phospho-NF-κB p65: r = 0.89, P < 0.001).The levels of phospho-NF-κB p65 protein were reduced in the nuclear fractions of the hearts of animals that received TDZD-8 (P < 0.05 vs. MI-R group), as shown in Fig. 3a.

a Representative Western Blot of an antibody against phospho- NF-κB p65 for each treatment group. b TLR2 mRNA was positively correlated with phospho-NF-κB p65 after MI-R in rats. These data obtained from quantitative densitometry are presented as mean ± SEM of n ≥ 6 animals per group. * P < 0.05 compared with shams; # P < 0.05 compared with MI-R animals
TDZD-8 attenuates myocardial infarct size
Ligation of the LAD for 30 min followed by 1 h reperfusion resulted in a substantial transmural infarct. The AAR expressed as a percentage of the left ventricular area (LV) was ~36%, and no difference was found among the MI-R and TDZD-8 groups (P > 0.05). The ratio of the infarct size (IS)/AAR was profound in the MI-R animals (35.62 ± 7.86% of AAR) (Fig. 4b). However, this ratio was reduced in the animals treated with TDZD-8 by nearly 33% compared with the MI-R animals (P < 0.05) (Fig. 4).

Infarct analysis after 1 h reperfusion in MI-R- and TDZD-8-treated rats. Percent of area at risk relative to LV (left) and infarcted area relative to AAR (right) in MI-R- and TDZD-8-treated rats. Each data point represents mean ± SEM of ≥6 animals from each group. # P < 0.05 vs. MI-R group
Discussion
The main findings of this study are: (1) TLR2/NF-κB signaling is significantly up-regulated following MI-R; (2) the levels of TLR2/NF-κB signaling agents are suppressed by treatment with TDZD-8; and (3) TDZD-8 treatment attenuates the progression of myocardial infarct size following MI-R. These findings suggest that GSK-3β inhibition attenuates MI-R-induced TLR2/NF-κB signaling that may facilitate development of secondary injury associated with reperfusion of the acutely ischemic myocardium.
TLR2-deficient mice have recently been shown to have less injured myocardium following I-R injury [6]. We have expanded on these results by defining the pattern of TLR2 mRNA and protein expression in MI-R model in rats, and by confirming a dominant role of innate immunity in ischemic cardiac injury. Also, we have demonstrated that TLR2 is constitutively expressed in hearts and the levels peak at 12 h after reperfusion. A more recent report by Favre et al. [5] showed that MI-R did not affect TLR2 gene expression (assessed by PCR). The inconsistency between our results is likely to be attributable to the difference in species used and the various experimental conditions, such as time of ischemia, duration of reperfusion, etc. We further analyzed heart-specific expression of TLR2 by immunohistochemistry (IHC) and found scattered foci of intense TLR2 staining involving two or more contiguous myocytes. When TLRs are stimulated by their specific ligands, they can activate NF-κB. After I-R injury, NF-κB activation is biphasic, with peaks after 15 min and 3 h attributed to the release of reactive oxygen species and the production of inflammatory cytokines, respectively. In this study, the activation of NF-κB was evaluated using an anti-phospho NF-κB p65 antibody that recognizes the activated nuclear form of NF-κB. We observed significant upregulation of positive NF-κB p65 cardiomyoctes after 1 h reperfusion. From these results, we may conclude that NF-κB activation is partially induced by TLR2. TLR2 and NF-κB p65 expression were markedly increased in the MI-R group, suggesting that this immune response is exacerbated following reperfusion. Persistent activation of NF-κB may cause excessive inflammatory cytokine release, culminating in tissue injury, organ dysfunction, or even death. In our results, we found that mRNA expression of TLR2 correlated significantly with cardiac levels of TNF-α and IL-6. Therefore, it is likely that activation of TLR2 by ligands released during MI-R injury may be key links between pro-inflammatory immune responses in the heart and myocardial damage and dysfunction.
Increased myocardial expression of TLR2/NF-κB after reperfusion injury may have evolved in a number of ways. It has been well established that oxidative stress in neonatal rat cardiomyocytes can activate NF-κB via TLR2 [12]. Increased cytokine levels may induce expression of TLR genes; conversely, tissues with higher TLR expression may be predisposed to increased cytokine production. This relationship needs to be further investigated. Necrotic cells have also been reported to modulate the expression of TLR2, however, the mechanism remains unclear [13].
Recently studies have shown the TLRs signaling can activate phosphatidylinositol 3-kinase (PI3K) [14], limiting the production of pro- and anti-inflammatory cytokines. These observations suggest that the role of PI3K in TLRs-mediated cytokine production is through inhibition of GSK-3β. GSK-3β has been shown to regulate NF-κB activation. GSK-3-null cells have diminished NF-κB activity [15] and pharmaceutical inhibitors of GSK-3β down-regulate NF-κB DNA binding activity [16]. These studies suggest that GSK-3β has the potential to modulate proinflammatory gene expression [17]. Martin et al. [9] indicated that GSK-3β inhibition by siRNA and certain pharmacological reagents increases IL-10 production but suppresses the release of pro-inflammatory cytokines. Furthermore, the ability of GSK-3β inhibition to reduce pro-inflammatory cytokines substantially, while augmenting IL-10 in response to a variety of TLR-agonists, demonstrates the broad ability of GSK-3β to attenuate the inflammatory response after TLR stimulation. In the present study, we found that GSK-3β inhibition by TDZD-8 decreased the mRNA level of TLR2 and acute phase cytokines. We also found that pretreatment with TDZD-8 significantly decreased phosphorylation of Ser536 on subunit p65 after 1 h of reperfusion. These data demonstrate that the ability of GSK-3β to regulate pro-inflammatory cytokines production is induced by TLR2 in cardiomyocytes. Shen et al. [18] have provided evidence that inhibition of GSK-3β by SB216763 or by overexpression of a dominant negative mutant of GSK-3β significantly enhanced TNF-α-expression in lipopolysaccharide-stimulated cardiomyocytes, in association with an increase in p65 phosphorylation. Our results are different from these observations since we used a chemical inhibitor. It is currently unclear whether this discrepancy results from different animal models used or the inhibition by chemical inhibitors is not entirely complete because of their effects on GSK-3β as chemical inhibitors often show nonselective activity.
Further research is needed to explore the pathways and mechanisms by which innate immunity is activated. The involvement of endogenous ligands and specific TLRs are not fully understood. Although the activation of TLR2 is deleterious to the injured myocardium, it must be taken into account that the same immune system is also involved in wound healing and tissue repair responses. For this reason, it represents a double-edged sword. Good timing in intervention can take away the ‘bad’ and enhance the ‘good’ in the inflammatory response during reperfusion.
Conclusion
In summary, to the best of our knowledge, this is the first study to demonstrate an effect of TLR2/NF-κB signaling in the injured heart after MI-R. In the present study, we found that MI-R up-regulated mRNA expression of TLR2 and acute phase cytokines, and NF-κB activity was increased in the heart adjacent to the injured site, which were all markedly inhibited by TDZD-8 administration. These results suggest that MI-R induces the activation of TLR2/NF-κB signaling, and this activation plays a central role in the inflammatory response that can further depress heart function following MI-R. The therapeutic benefit of GSK-3β inhibition after MI-R may be due to its effect on modulating TLR2/NF-κB signaling.
References
Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9.
Aliprantis AO, Yang RB, Weiss DS, Odowski PG, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J 2002;19:3325–36.
Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–803.
Ziegler G, Harhausen D, Schepers C, Hoffmann O, Röhr C, Prinz V, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–9.
Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, et al. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1064–71.
Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, et al. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292(1):H503–9.
Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2- knockout mice. Circulation. 2004;110(18):2869–74.
Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–8.
Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6(8):777–84.
Tanno M, Tsuchida A, Nozawa Y, Matsumoto T, Hasegawa T, Miura T, et al. Roles of tyrosine kinase and protein kinase C in infarct size limitation by repetitive ischemic preconditioning in the rat. J Cardiovasc Pharmacol. 2000;35(3):345–52.
Cuzzocreas S, Mazzon E, Esposito E, Muia C, Abdelrahman M, Paola RD, Crisafulli C, Bramanti P, Thiemermann C. Glycogen synthase kinase-3beta inhibition attenuates the development of ischaemia/reperfusion injury of the gut. Intensive Care Med. 2007;33(5):880–93.
Frantz S, Kelly RA, Bourcier T. Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem. 2001;276(7):5197–203.
Lee H, Jo EK, Choi SY, Oh SB, Park K, Kim JS, et al. Necrotic neuronal cells induce inflammatory Schwann cell activation via TLR2 and TLR3: implication in Wallerian degeneration. Biochem Biophys Res Commun. 2006;350(3):742–7.
Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull. 2007;30(9):1617–23.
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90.
Demarchi F, Bertoli C, Sandy P, Schneider C. Glycogen synthase kinase-3 beta regulates NF-kappa b1/p105 stability. J Biol Chem. 2003;278:39583–90.
Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock. 2007;27(2):113–23.
Shen E, Fan J, Peng T. Glycogen synthase kinase-3beta suppresses tumor necrosis factor-alpha expression in cardiomyocytes during lipopolysaccharide stimulation. J Cell Biochem. 2008;104(1):329–38.
Acknowledgment
The authors thank Dr. Ling Tao (Department of Cardiology, Xi Jing Hospital, Fourth Military Medical University) for her help and careful revision of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: S. Stimpson.
Rights and permissions
About this article
Cite this article
Gao, HK., Yin, Z., Zhang, RQ. et al. GSK-3β inhibitor modulates TLR2/NF-κB signaling following myocardial ischemia-reperfusion. Inflamm. Res. 58, 377–383 (2009). https://doi.org/10.1007/s00011-009-0002-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-009-0002-1