
Abstract
Vitamin E succinate (VES), a derivative of vitamin E, is a promising cancer chemopreventive agent that inhibits tumor promotion by inducing apoptotic cell death. The effects of VES on autophagy, an intricate programmed process which helps cells survive in some stressed situations by degrading some cytoplasmic material, are unclear. When human gastric cancer cells SCG-7901 were exposed to VES, both the level of microtubule-associated protein 1 light chain 3 and the yeast ATG6 homolog Beclin-1 increased, and related autophagy genes were activated, thereby suggesting that autophagy was induced by VES. We also observed that VES-induced autophagy was accompanied by the activation of AMP-activated protein kinases (AMPK). VES-induced autophagy decreased when AMPK was inhibited by using small interfering RNA (siRNA), thereby suggesting that VES-induced autophagy is mediated by AMPK. Moreover, further studies revealed that the decreased activity of mammalian target of rapamycin (mTOR) and its downstream targets P70S6K and 4EBP-1 were involved in VES-activated autophagy associated with AMPK activation. The experiments also showed that the activity of protein kinases B (Akt)-mTOR axis was inhibited by VES. VES-induced AMPK activation could be attenuated by Akt activation. Overall, our studies demonstrated that AMPK was involved in the VES-induced autophagy. Crosstalk exists between AMPK and the Akt/mTOR axis. The results elucidated the mechanism of VES-induced autophagy in human gastric cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is the third leading cause of cancer death worldwide, and approximately 700,000 cases of stomach cancer are diagnosed each year [1]. Chemotherapy is the major clinical treatment for advanced gastric cancer, but the development of effective chemotherapeutic drug is rather slow. Drug resistance and the toxicity of chemotherapy are great limitations to this kind of treatment [2]. Therefore, the study of new treatments that overcome the aforementioned drawbacks is essential.
Vitamin E succinate (VES; α-tocopherol succinate), a vitamin E derivative, has potent anticancer effects on a variety of cancers. Previous research has shown that VES can induce human gastric cancer SGC-7901 cell apoptosis through many signaling pathways, such as extrinsic Fas, endoplasmic reticulum stress, and mitogen-activated protein kinase (MAPK) pathways [3–5]. VES, which has anticancer effects, shows no toxicity to normal cells and tissues in vitro and in vivo [6–12]. However, the relationship between VES and autophagy and the underlying molecular mechanisms is poorly defined.
Autophagy is a dynamic and highly regulated process that plays a critical role in degradation of long-lived proteins, bulk cytoplasm, and organelles [13–16]. This process begins with the formation of double-membrane autophagosomes initiated by PI3 kinase (PI3K) type III-Atg6/Beclin-1 cascade [17]. After the formation of autophagosomes, two ubiquitin-like systems exist: the Atg8/LC3-phosphatidylethanolamine conjugate system and the Atg12–Atg5 conjugate system. Both systems are involved in the expansion of the autophagosomes [18]. Autophagy is regulated by numerous cross talking signaling pathways, which function in response to many forms of cellular stress, such as hypoxia, starvation, and radiation [19]. The classic PI3K/Akt-mTOR signaling pathway is considered as a crucial negative regulator for the formation of autophagosomes [20]. mTOR activity is controlled by PI3K/Akt signaling pathway. Activated Akt phosphorylates tuberous sclerosis complex 2 (TSC2) prevents formation of the inhibitory TSC1/TSC2 heterodimer and then activates TOR complex 1 (TORC1), which is a functionally distinct complex of mTOR. When mTOR is activated, it stimulates protein synthesis by phosphorylating key translation regulators, i.e., ribosomal protein S6 kinase (P70S6K) and eukaryotic initiation factor 4E binding protein 1 (4EBP-1) [21, 22]. Recent studies have shown that AMPK activation may lead to autophagy through the negative regulation of mTOR [23–26]. Activated AMPK inhibits TORC1 activity by phosphorylation and activation of the negative regulator TSC2 [27]. AMPK can directly phosphorylate ULK1 (Unc-51-like kinase 1) to induce autophagy [28]. In the present study, we investigated the cellular responses of autophagy process induced by VES in human gastric cancer cells, and whether the accumulation of AMPK was involved in these processes. We also probed whether crosstalk is present between AMPK and Akt/mTOR signaling pathways. Our studies would provide a better understanding for the implication of autophagy in VES-mediated anticancer activities.
Materials and methods
Cells and cell culture
Human gastric cancer SGC-7901 cells were obtained from Chinese Academy of Sciences Cell Resource Center. Cells were maintained in RPMI1640 medium (Hyclone, SH30809.01B) supplemented with 10% fetal bovine serum (FBS) (SiJiQing, 13011-8611), 10,000 IU penicillin, and 10,000 μg/mL streptomycin. The cells were cultured in a humidified 5% CO2 atmosphere at 37 °C.
Chemical treatment and antibodies
VES (Sigma, T3126) dissolved in sterile absolute ethanol; 3-methyladenine (3-MA) (Sigma, M9281), chloroquine (CQ) (Sigma, C6628), and IGF-1 (Abcam, ab123776) dissolved in phosphate-buffered saline (PBS); AICAR (Sigma, A9978), LY294002 (Sigma, L9908), Compound C (Sigma, P5499), and Rapamycin (RAPA) (Sigma, 37094) dissolved in dimethyl sulfoxide (DMSO) were added to the medium to final concentrations, as described in each experiment. Cells grown to 60% confluency were switched to 2% FBS medium, and the culture was allowed to expand for 24 h before any treatment was performed. For 3-MA, CQ, RAPA, AICAR, LY294002, Compound C, and IGF-1 plus VES treatment, cells were first incubated with 5 mM 3-MA, 20 μM CQ, 100 nM RAPA, 1 mM AICAR, 10 μM LY294002, and 10 μM Compound C for 2 h, respectively. Then, VES was added, and the cells were incubated for 24 h. For IGF-1 plus VES treatment, cells were pretreated with 100 ng/mL IGF-1 for 1 h and then incubated with VES for 24 h.
Antibodies were obtained from the following sources: AMPKα (Cell signaling, 5831S), p-AMPKα (Thr172) (Cell signaling, 2535S), ACC (Cell signaling, 3662), p-ACC (Ser97) (Cell signaling, 11818), Akt (Cell signaling, 9272), p-Akt (S473) (Cell signaling, 9271S), mTOR (Cell signaling, 2972S), p-mTOR (Ser2448) (Cell signaling, 5536S), P70S6K (Cell signaling, 5707S), p-P70S6K (T389) (Cell signaling, 9205S), 4EBP-1 (Cell signaling, 8594S), p-4EBP-1 (T37/46) (Cell signaling, 9459S), LC3 (Sigma, L7543), Beclin-1 (Cell signaling, 3495), β-actin (Santa Cruz, sc-1616), Alkaline phosphatase-conjugated second antibody anti-rabbit IgG (Promega, S373B), and Fluorescein-Conjugated AffiniPure Goat anti-Rabbit IgG (H+L) (ZSGB-BIO, ZF-0311).
Western blot analyses
The cells were lysed in the RIPA buffer mixed with Phosphatase Inhibitors Cocktail (Roche, 04906845001) or Protease Inhibitors (Roche, 11206893001). After incubation for 30 min at 4 °C, the sample was centrifuged at 15,000×g for 8 min at 4 °C, and the supernatant was collected as whole cell lysate. Equivalent amounts of proteins were separated on 10% SDS–PAGE gels and transferred to a polyvinylidene difluoride membrane (Merck Millipore, IPFL00010). The blots were blocked in 5% (w/v) bovine serum albumin (BSA) and incubated with a 1:1000 dilution of primary antibodies overnight at 4 °C, followed by incubation with goat anti-rabbit alkaline phosphatase-conjugated IgG secondary antibody. The bolts were then detected by using the Western Blue Stabilized Substrate for alkaline phosphatase (Promega, S3841). The blots were analyzed by using Chemilmager 4000 (Alpha Innotech, USA).
Quantitative real-time PCR
Total RNA was isolated with Trizol reagent (Invitrongen, 10296010) according to the manufacturer’s instructions. Total RNA at 1 μg was reverse-transcribed into cDNA with High-Capacity cDNA Reverse Transcription Kit (Invitrongen, 4368813). For Real-time PCR, 1 μL of the cDNA was performed in triplicate with SYBR® Select Master Mix (Invitrongen, 4472980) for 10 min at 95 °C for initial denaturation, followed by 40 cycles of segments of 95 °C for 15 s and 60 °C for 60 s in the 7500 Fast real-time PCR system (Applied Biosystems). Primers were designed using Primer Premier 5 software. The primer sequences and the expected length of PCR products are shown in Table 1. The expression levels of the LC3, Beclin-1, and AMPK genes were normalized against the expression levels of the housekeeping gene β-actin.
RNA interference
Small interfering AMPK RNA sequences (5′-UGG CAA ACA GGU UGA AUU Gtt-3′) and negative control siRNA were purchased in Ambion. When the cells were grown to 40–50% confluency, Opti-MEM I Reduced Serum Medium was used to suspend the cells. The siRNA at 100 pmol was added to 250 μL Opti-MEM I Reduced Serum Medium. Two copies of the 5 μL Lipofectamine 2000 reagent (Invitrogen, 11668027) were added to 250 μL Opti-MEM I Reduced Serum Medium. The above-mentioned siRNA-containing solution and the Lipofectamine 2000 solution were mixed and added to cells. After incubation for 6 h, the conventional culture solution was changed with 10% RPMI-1640 medium. Then, cells were subjected to 20 μg/mL VES for another 24 h.
Immunocytochemistry
After treatment, human gastric cancer SGC-7901 cells were fixed in 4% paraformaldehyde (Thermo Fisher, NC9245948), perforated with 0.5% Triton X-100 (BIOSHARP, 0694), and then blocked with 1% bovine serum albumin (Roche, 735094) in PBS. Cells were stained with rabbit anti-LC3 (1:200) primary antibody, visualized using Fluorescein-Conjugated AffiniPure Goat anti-Rabbit IgG (H+L), and counterstained with DAPI dihydrochloride (Sigma, 30670). These fluorescent-stained cells were observed under a confocal microscope (Nikon, Japan). Final images were a non-contrast-adjusted merge of two channels. The number of LC3-positive dots per cell was quantified by Image-Pro Plus. At least ten cells in two independent experiments were analyzed randomly.
Statistical analysis
Multiple comparisons were analyzed using one-way analysis of variance (ANOVA), and individual differences were tested by the Fisher’s protected least-significant difference test after demonstrating the existence of significant intergroup difference by one-way ANOVA. Two-group comparisons were performed using Student’s t test. Results are shown as mean ± SD values, with a minimum of three separate experiments for each issue addressed. p values of less than 0.05 were considered significant.
Result
VES-induced autophagy in SGC-7901 cells
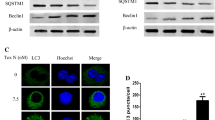
We investigated whether VES could elicit autophagy in SGC-7901 cells by Western blot analysis. As shown in Fig. 1a, significantly increased protein expressions of LC3 (LC3-II) and Beclin-1 were observed in SGC-7901 cells treated with VES in both dose- and time-dependent manners. VES-induced cleavage of LC3 and the increased expression of Beclin-1 were attenuated by the treatment of 5 mM 3-MA. Then, the mRNA levels of LC3 and Beclin-1 were also assessed with quantitative Real-time PCR. As shown in Fig. 1b, both the levels of mRNA of LC3 and Beclin-1 significantly increased after VES treatment at 20 μg/mL when compared with the control group.

Autophagy is induced in SGC-7901 cells after VES treatment. a Protein levels of LC3 and Beclin-1 were measured using immunoblot analysis. SGC-7901 cells were treated with various concentrations of VES for 24 h and 20 μg/mL VES treated for different times. In 3-MA plus VES treatment, cells were first incubated with 5 mM 3-MA for 2 h. RAPA (100 nM) as a positive control. b LC3 and Beclin-1 mRNA expressions in response to VES were examined by quantitative Real-time PCR analysis. c Representative images of LC3 fluorescent staining in SGC-7901 cells with or without VES treatment. Cells were treated with different doses of VES for 24 h. DMOS (<0.1%) as a negative control. (a) DMSO; (b) 5 μg/mL VES; (c) 10 μg/mL VES; (d) 20 μg/mL VES; (e) 3-MA+VES. d Immunoblot analysis of LC3 expression from lysates of SGC-7901 cells treated with various concentrations of VES for 24 h in the absence or presence of lysosomal inhibitors (20 μM CQ), *compared with control p < 0.05, #compared with the group of 20 μg/mL VES, p < 0.05
Furthermore, the immunofluorescence technique was used to observe the LC3-positive fluorescence spots and distribution of LC3 in the SGC-7901 cells after the VES treatment. Negative control failed to show LC3 fluorescent spots, whereas the LC3 fluorescent spots aggregation gradually increased with increasing VES concentration; 3-MA can significantly weaken the VES-induced LC3-positive spots (Fig. 1c).
The role of VES is related to initiation of autophagy instead of the inhibition of lysosome degradation. SGC-7901 cells were co-treated with CQ and VES to further prove that autophagy could be triggered by VES. LC3-II accumulated with increasing VES dose. When cells were co-treated with CQ and VES, the expression of LC3-II was higher than when CQ was absent (Fig. 1d).
VES treatment increased the phosphorylation of AMPK and its substrate
The AMPK subunits were examined to define their roles on VES-treated SGC-7901 cells; AICAR, as an adenosine analog to activate AMPK, was used as a positive control in this set-up. As shown in Fig. 2, both phosphorylations of AMPKα at Thr172 and the AMPK substrate acetyl-CoA carboxylase (ACC) increased upon VES treatment in both dose- and time-dependent manners. AMPK was involved in VES-induced autophagy.

Increased expression of AMPK in response to VES. a Dose-dependent analysis of p-AMPK, AMPK, p-ACC, and ACC in SGC-7901 cells. Cells were exposed to VES at the indicated concentrations for 24 h. AICAR at 1 mM was used as a positive control. b Time-dependent analysis of p-AMPK, AMPK, p-ACC, and ACC in SGC-7901 cells. Cells were treated with 20 μg/mL VES for the indicated time periods, * and # p < 0.05 compared to control
Silencing AMPK attenuates VES-induced autophagy
To further define the role of AMPK in VES-induced autophagy, AMPK was silenced by siRNA method. The transfection efficiency is shown in Fig. 3a. The reduction of LC3 and Beclin-1 expressions demonstrated that VES-mediated autophagy was inhibited (Fig. 3b). Notably, AMPK silencing by siRNA also reduced the mRNA expression of LC3 (Fig. 3c). In addition, we also used immunofluorescence to observe the LC3-positive fluorescence dots and distribution of LC3, and the result showed that the LC3-positive dots induced by VES was significantly weakened when AMPK was silenced (Fig. 3d). Collectively, these results suggest that VES signals autophagy in SGC-7901 cells via the activation of AMPK.

AMPK-mediated VES-induced autophagy. a Transfection efficiency. Cells were transfected with negative control or AMPK-siRNA for 48 h. Then, the mRNA expression of AMPK was detected by quantitative Real-time PCR and protein level of AMPK was measured by immunoblot analysis. b Cells were transfected with AMPK-siRNA for 48 h and were treated with 20 μg/mL VES for 24 h. Western blot was used to measure the protein expressions of LC3 and Beclin-1. c Cells were treated with the same method as B. Quantitative Real-time PCR was used to measure the mRNA expression of LC3. d Cells were treated with the same method as B. Immunofluorescence was used to observe the fluorescence dots of LC3. 1 Negative control. 2 AMPK-siRNA. 3 20 μg/mL VES. 4 AMPK-siRNA plus 20 μg/mL VES. *Compared with control p < 0.05, #compared with the group of VES alone, p < 0.05
Akt-mTOR signaling pathway was inhibited by VES
We examined whether Akt and mTOR phosphorylations were involved in VES-induced autophagy in SGC-7901 cells. As shown in Fig. 4a, both Akt (S473) and mTOR (S2448) phosphorylations were inhibited by VES treatment in SGC-7901 cells. In addition, the dephosphorylations of Akt and mTOR mediated by VES relied on the increased doses of VES. The phosphorylation status of both P70S6K (T389) and 4EBP-1 (T37/46) were also examined with immunoblot analysis. As shown in Fig. 4b, the VES-treated dephosphorylations of both P70S6K (T389) and 4EBP-1 (T37/46) were in a dose-dependent manner.

VES suppressed the Akt/mTOR signaling pathway. Immunoblot analysis of both total and phosphorylation levels of Akt (S473), mTOR (S2448), P70S6K (T389), and 4EBP-1 (T37/46) from lysates of SGC-7901 cells treated with various concentrations of VES for 24 h, * and # p < 0.05 compared to respective treatment group in control medium
mTOR signaling was inhibited by VES through AMPK activation
To define whether mTOR phosphorylation was regulated by VES-activated AMPK, the cells were treated with AMPK activator AICAR or inhibitor compound C before VES treatment. The p-AMPK was significantly activated when cells were treated with 1 mM AICAR or AICAR plus VES. However, the phosphorylations of mTOR, P70S6K, and 4EBP-1 decreased (Fig. 5a). AMPK was inhibited when the cells were administered with 10 μM Compound C. The p-AMPK expression was significantly inhibited. The phosphorylations of mTOR, P70S6K, and 4EBP-1 were clearly increased. The p-AMPK was weakened when co-treated with Compound C and VES, compared with VES treatment. Meanwhile, the p-mTOR, p-P70S6K, and p-4EBP-1 were enhanced (Fig. 5b). When the AMPK was silenced with transfecting siRNA before VES treatment, the results showed that both p-AMPK and total AMPK were significantly inhibited, but the phosphorylations of mTOR, p70S6K, and 4EBP-1 increased (Fig. 5c).

VES inhibited mTOR signaling through AMPK activation. a AMPK activation inhibited mTOR-related signaling. SGC-7901 cells were incubated in the presence or absence of 1 mM AICAR for 2 h, and then were treated with 20 μg/mL VES for 24 h. After cell lysis, both the total and phosphorylation levels of AMPK, mTOR, P70S6K, and 4EBP-1 were analyzed by protein gel blotting. b AMPK inhibition activated mTOR-related signaling. Cells were incubated in the presence or absence of 10 μM Compound C for 2 h and then were treated with 20 μg/mL VES for 24 h. Protein gel blotting was used to measure both the total and phosphorylation levels of AMPK, mTOR, P70S6K, and 4EBP-1. c AMPK silencing activated mTOR-related signaling. Cells were transfected with control siRNA or AMPK-siRNA for 48 h and then treated with VES for 24 h. Protein expressions of total and phosphorylation levels of AMPK, mTOR, P70S6K, and 4EBP-1 were analyzed by protein gel blotting
AMPK was activated by VES through the inhibition of Akt
To test whether Akt signaling was involved in AMPK activation, the expression of p-AMPK, AMPK, p-Akt, and Akt were determined in SGC-7901 cells treated with 100 ng/mL Akt activator IGF-1 or 10 μM Akt inhibitor LY294002. The increased p-Akt activation and simultaneously decreased p-AMPK activation were observed when the cells were treated with IGF-1. Meanwhile, the VES-attenuated p-Akt activation was increased and VES-induced p-AMPK activation was decreased (Fig. 6a). On the contrary, when cells were treated with LY294002, lower p-Akt activation and higher p-AMPK activation compared with control were revealed. In addition, more obvious decrease of p-Akt activation and increase of p-AMPK activation were observed under combined treatment of VES and LY294002 (Fig. 6b).

VES-activated AMPK through Akt inhibition. a Akt activation inhibited AMPK signaling. SGC-7901 cells were incubated in the presence or absence of 100 ng/mL IGF-1 for 2 h and then were treated with 20 μg/mL VES for 24 h. After cell lysis, both the total and phosphorylation levels of Akt and AMPK were analyzed by protein gel blotting. b Akt inhibition activated AMPK signaling. Cells were incubated in the presence or absence of 10 μM LY294002 for 2 h, and then treated with 20 μg/mL VES for 24 h. Protein gel blotting was used to measure the expression of Akt, p-Akt, AMPK, and p-AMPK, * and # p < 0.05 compared to control
Discussion
VES, a vitamin E analog, is a promising chemoprevention agent used in a variety of cancer models [29–32]. VES exhibits potent anti-proliferative effects on human tumor cells and selectively induces apoptosis of tumor cells in vitro and in vivo, but not of normal cells [31, 33]. A recent study also showed that VES induced autophagy in human gastric cancer cells [34]. Consistent with previous reports, our study showed that VES induced autophagy in gastric cancer cells, and AMPK played an important role in VES-induced autophagy. (Fig. 7).

Schematic model illustrating the potential pathway associated with VES-induced autophagy in SGC-7901 cells. VES-induced autophagic activation is mediated by the Akt/AMPK/mTOR pathway. VES causes the phosphorylation of AMPK at Thr172 and activates AMPK by decreasing the phosphorylation of Akt. This leads to inhibition of mTOR signaling and its downstream tartrates P70S6 K and 4EBP-1, thereby negatively regulating autophagy. Silencing of AMPK by using siRNA could markedly attenuate VES-induced autophagy
Autophagy is a highly regulated process of sequestrating cytoplasmic proteins into lytic compartments and is characterized by the formation of the autophagosome, which then fuses with lysosomes where the contents are degraded [35, 36]. The formation of autophagosome was initiated by PI3 kinase type III-Atg6/Beclin-1 cascade and was expanded by Atg8/LC3-phosphatidylethanolamine conjugate system. Accumulating anticancer agents have been documented to trigger the cellular autophagic process, but the signaling pathway involved in autophagy is still controversial. In this study, we detect the regulatory effect of VES on autophagy in human gastric cancer SGC-7901 cells. The results indicated that VES is a strong inducer of autophagy, as evidenced by the significant increase of LC3 and Beclin-1 expressions. LC3 is a homolog of yeast Atg8 and is generally used as a marker protein for tracing the autophagic process. LC3 is post-translationally modified by an ubiquitination-like reaction and cleaved off to assume a soluble form, LC3-I, thereby exposing its carboxyl terminal glycine. Upon autophagy induction, LC3-I is modified with phosphatidylethanolamine at the glycine residue and becomes LC3-II, which is bound to both the outer and inner membranes of the autophagosome. Therefore, the formation of LC3-II is a good marker to monitor the occurrence of autophagosome formation [37]. Beclin-1, as a key molecule, is involved in the formation of autophagosome by PI3 kinase type III-Atg6/Beclin-1 cascade. Our study demonstrated that VES treatment increased both LC3 and Beclin-1 expressions in dose- and time-dependent manners in SGC-7901 cells. In addition, the mRNA of LC3 and Beclin-1 also increased after VES treatment. The immunofluorescence study also supports this conclusion.
The involved mechanisms in autophagy regulation are multifactorial. Among these, AMPK integrates input information from multiple upstream signal transduction pathways and has been well identified as a vital positive regulator in autophagy. AMPK also serves as a key regulator of cell survival or death in response to various pathological stresses, including oxidative stress, endoplasmic reticulum stress, hypoxia, and osmotic stress [38, 39]. AMPK activation can regulate the tumor suppressor gene P53, mTOR, ULK1, and other downstream signal molecular activities involved in autophagy [28, 40, 41]. As an elementary mechanism, autophagy promotes cell survival under stress situations. Emerging evidence has presented the critical role of AMPK axis on autophagy [42]. Therefore, we hypothesize that AMPK would be involved in autophagy activation induced by VES in human gastric cancer cells. In the present study, we found that VES could activate AMPK in cultured SGC-7901 cells in a dose- and time-dependent manners. Increased AMPK activity upon VES treatment was further proven by analysis of the phosphorylation of the AMPK substrate acetyl-CoA carboxylase. Additionally, AMPK inhibition by siRNA significantly reduced the protein and mRNA expressions of LC3 and Beclin-1. Immunofluorescence study also showed that silencing AMPK by siRNA significantly weakened the VES-induced fluorescence dots of LC3, which suggested that VES-mediated autophagy was inhibited. All of the above-mentioned results demonstrated that the AMPK signaling pathway is involved in VES-induced autophagy.
The Akt-mTOR signaling pathway is the major negative signaling pathway against autophagy [43]. The serine/threonine protein kinase Akt mediates mTOR activity [44] and positively regulates mTOR, which is a major component of autophagy regulation. mTOR phosphorylation increases the levels of downstream targets, such as P70S6K and 4EBP-1, to regulate many different cellular processes [45, 46]. The P70S6K phosphorylation level is important in the initiation of the translation of proteins associated with cell growth and proliferation [47]. Our data demonstrated that VES dephosphorylated Akt and mTOR as well as P70S6K and 4EBP-1 (the two best-characterized targets of mTOR1 complex) in gastric cancer cells, thereby emphasizing the functional importance of the involved Akt-mTOR signaling in VES-mediated autophagy in gastric cancer cells.
The signaling by which VES induces autophagy has been suggested to be a direct action of mTOR through AMPK activation [34]. As a central signal integrator, mTOR receives signals arising from nutrients, growth factors, and many cellular kinases including AMPK [40, 48]. Phosphorylation of AMPK activates downstream signaling that leads to mTOR inhibition and triggers autophagy [49], which is consistent with the AMPK function of initiating catabolic processes [25]. Our data showed that the activity of mTOR and its downstream targets, including P70S6 K and 4EBP-1, was inhibited after AMPK activation because of AICAR treatment. On the contrary, pre-treatment with AMPK inhibitor Compound C could attenuate the VES-induced inhibition of mTOR and its downstream targets. Then, AMPK silencing also weakened VES-induced inhibition of mTOR. Therefore, VES-induced AMPK activation prohibits mTOR signaling, and then triggers the initiation of autophagy.
Akt has been shown to be an effective cellular antagonist of AMPK [50–52]. It has been reported that high glucose suppresses AMPK T172 phosphorylation through Akt-dependent phosphorylation of AMPK S485, which is a function to inhibit AMPK activation by multiple stimulations [53]. Our study also highlights the importance of the crosstalk between AMPK and Akt. When cells were treated with Akt activator IGF-1, the VES-induced AMPK activation was inhibited. On the contrary, for cells treated with Akt inhibitor LY294002, the VES-induced AMPK activation strengthened.
This study had some limitations. The relationship between VES-induced autophagy and the effect of VES to suppress tumor growth have not been explored. Therefore, the significance of this autophagy is unknown in terms of the VES anticancer effect. Moreover, autophagy flux should be detected when AMPK was inhibited or activated by Compound C or AICAR. However, autophagy may be influenced by Compound C or AICAR through other signaling pathways besides AMPK axis.
Collectively, this study highlights the significance of AMPK involved in VES-induced autophagy in SGC-7901 cells. VES initially triggers an AMPK-mediated autophagic process via Akt/AMPK/mTOR signaling. Information provided in this paper would be valuable for further studies. This study also contributes to explore the underlying mechanisms of VES-induced autophagy in human gastric cancer cells.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65(2):87–108
Ohtsu A (2008) Chemotherapy for metastatic gastric cancer: past, present, and future. J Gastroenterol 43(4):256–264
Huang X, Zhang Z, Jia L, Zhao Y, Zhang X, Wu K (2010) Endoplasmic reticulum stress contributes to vitamin E succinate-induced apoptosis in human gastric cancer SGC-7901 cells. Cancer Lett 296(1):123–131
Wu K, Li Y, Zhao Y, Shan YJ, Xia W, Yu WP, Zhao L (2002) Roles of Fas signaling pathway in vitamin E succinate-induced apoptosis in human gastric cancer SGC-7901 cells. World J Gastroenterol 8(6):982–986
Wu K, Zhao Y, Li GC, Yu WP (2004) c-Jun N-terminal kinase is required for vitamin E succinate-induced apoptosis in human gastric cancer cells. World J Gastroenterol 10(8):1110–1114
Israel K, Yu W, Sanders BG, Kline K (2000) Vitamin E succinate induces apoptosis in human prostate cancer cells: role for Fas in vitamin E succinate-triggered apoptosis. Nutr Cancer 36(1):90–100
Neuzil J, Weber T, Gellert N, Weber C (2001) Selective cancer cell killing by α-tocopheryl succinate. Br J Cancer 84(1):87–89
Sahu SN, Edwards-Prasad J, Prasad KN (1988) Effect of alpha tocopheryl succinate on adenylate cyclase activity in murine neuroblastoma cells in culture. J Am Coll Nutr 7(4):285–293
Wu K, Zhao Y, Liu BH, Li Y, Liu F, Guo J, Yu WP (2002) RRR-alpha-tocopheryl succinate inhibits human gastric cancer SGC-7901 cell growth by inducing apoptosis and DNA synthesis arrest. World J Gastroenterol 8(1):26–30
Yu W, Israel K, Liao QY, Aldaz CM, Sanders BG, Kline K (1999) Vitamin E succinate (VES) induces Fas sensitivity in human breast cancer cells: role for Mr 43,000 Fas in VES-triggered apoptosis. Cancer Res 59(4):953–961
Zhang X, Peng X, Yu W, Hou S, Zhao Y, Zhang Z, Huang X, Wu K (2011) Alpha-tocopheryl succinate enhances doxorubicin-induced apoptosis in human gastric cancer cells via promotion of doxorubicin influx and suppression of doxorubicin efflux. Cancer Lett 307(2):174–181
Bang OS, Park JH, Kang SS (2001) Activation of PKC but not of ERK is required for vitamin E-succinate-induced apoptosis of HL-60 cells. Biochem Biophys Res Commun 288(4):789–797
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A (2009) Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 16(7):966–975
Gozuacik D, Kimchi A (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23(16):2891–2906
Klionsky DJ (2010) The autophagy connection. Dev Cell 19(1):11–12
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12(9):814–822
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9(10):1102–1109
Geng J, Klionsky DJ (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 9(9):859–864
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40(2):280–293
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22(2):124–131
Asnaghi L, Bruno P, Priulla M, Nicolin A (2004) mTOR: a protein kinase switching between life and death. Pharmacol Res 50(6):545–549
Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K (2008) Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 183(1):101–116
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, Jaattela M (2009) TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J 28(6):677–685
Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ (2006) AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 281(46):34870–34879
Papandreou I, Lim AL, Laderoute K, Denko NC (2008) Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ 15(10):1572–1581
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115(5):577–590
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141
Neuzil J, Weber T, Gellert N, Weber C (2001) Selective cancer cell killing by alpha-tocopheryl succinate. Br J Cancer 84(1):87–89
Wu K, Shan YJ, Zhao Y, Yu JW, Liu BH (2001) Inhibitory effects of RRR-alpha-tocopheryl succinate on benzo(a)pyrene (B(a)P)-induced forestomach carcinogenesis in female mice. World J Gastroenterol 7(1):60–65
Neuzil J, Weber T, Schroder A, Lu M, Ostermann G, Gellert N, Mayne GC, Olejnicka B, Negre-Salvayre A, Sticha M, Coffey RJ, Weber C (2001) Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J 15(2):403–415
Sun Y, Zhao Y, Hou L, Zhang X, Zhang Z, Wu K (2014) RRR-alpha-tocopheryl succinate induces apoptosis in human gastric cancer cells via the NF-kappaB signaling pathway. Oncol Rep 32(3):1243–1248
Weber T, Lu M, Andera L, Lahm H, Gellert N, Fariss MW, Korinek V, Sattler W, Ucker DS, Terman A, Schroder A, Erl W, Brunk UT, Coffey RJ, Weber C, Neuzil J (2002) Vitamin E succinate is a potent novel antineoplastic agent with high selectivity and cooperativity with tumor necrosis factor-related apoptosis-inducing ligand (Apo2 ligand) in vivo. Clin Cancer Res 8(3):863–869
Hou L, Li Y, Song H, Zhang Z, Sun Y, Zhang X, Wu K (2015) Protective macroautophagy is involved in vitamin e succinate effects on human gastric carcinoma cell line SGC-7901 by inhibiting mTOR axis phosphorylation. PLoS ONE 10(7):e0132829
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19(21):5720–5728
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152(4):657–668
Lin H-Y, Lin J-N, Ma J-W, Yang N-S, Ho C-T, Kuo S-C, Way T-D (2015) Demethoxycurcumin induces autophagic and apoptotic responses on breast cancer cells in photodynamic therapy. J Funct Foods 12:439–449
Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ (2000) Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 49(4):527–531
Hoyer-Hansen M, Jaattela M (2007) AMP-activated protein kinase: a universal regulator of autophagy? Autophagy 3(4):381–383
Alers S, Loffler AS, Wesselborg S, Stork B (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32(1):2–11
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10(6):676–687
Carling D (2004) The AMP-activated protein kinase cascade–a unifying system for energy control. Trends Biochem Sci 29(1):18–24
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306(5698):990–995
Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18(16):1926–1945
Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schafer BW, Schrappe M, Stanulla M, Bourquin JP (2010) Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Investig 120(4):1310–1323
Feng Z, Zhang H, Levine AJ, Jin S (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102(23):8204–8209
Pouyssegur J, Dayan F, Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441(7092):437–443
Faivre S, Kroemer G, Raymond E (2006) Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov 5(8):671–688
Shaw RJ (2006) Glucose metabolism and cancer. Curr Opin Cell Biol 18(6):598–608
Berggreen C, Gormand A, Omar B, Degerman E, Goransson O (2009) Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J physiol Endocrinol Metab 296(4):E635–E646
Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N (2005) Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem 280(37):32081–32089
Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281(9):5335–5340
Ning J, Xi G, Clemmons DR (2011) Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 152(8):3143–3154
Funding
This study was funded by a Grant from the National Natural Science Foundation of China (No. 81172651) to K. Wu.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Yu, Y., Hou, L., Song, H. et al. Akt/AMPK/mTOR pathway was involved in the autophagy induced by vitamin E succinate in human gastric cancer SGC-7901 cells. Mol Cell Biochem 424, 173–183 (2017). https://doi.org/10.1007/s11010-016-2853-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-016-2853-4