
Abstract
In the current work, the thermal curing process of maleinated acrylated epoxidized linseed oil (MaAELO) mixed with reactive diluent, initiator, and accelerator was studied by using isothermal differential scanning calorimetry (DSC) and dynamic rheology. The investigations encompassed (1) the determination of the whole set of apparent kinetic parameters, Ea(α), A(α), and f(α) by using two accurate isoconversional kinetic analysis methods and the compensation effect, (2) the prediction of cure conversion curves, α(t), at arbitrary processing temperatures with all three kinetic parameters in comparison with the prediction based on Ea(α) alone, and (3) the determination of molecular and macroscopic gelation during the thermal cure of MaAELO resin mixture. The thermal cure of MaAELO resin mixture did not start immediately but after a remarkable induction period. It was possible to determine the kinetic parameters and use them to predict the induction period for arbitrary process temperature. In addition, the glass transition temperature, Tg, of thermally cured MaAELO resin mixture was measured by thermomechanical analysis and dynamic DSC.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past decades, there has been a growing interest in the use of polymers based on renewable resources because of diminishing petroleum resources and environmental issues. Vegetable oils are triglyceride oils based on different fatty acids that are responsible for making them a versatile natural and renewable resource for a variety of polymeric materials [1]. The routes to synthesize unsaturated polyester resins (UPR) from a broad range of renewable resources including vegetable oils have been reviewed recently by Ma et al. [2] and in more detail, including the synthesis routes to achieve bio-based reactive diluents for UPR, by Li et al. [3]. In the current work, we followed the synthesis route of epoxidation of linseed oil (ELO) which was acrylated (AELO) afterward and finally reacted with Maleic anhydride (MaAELO). Wool and his team first proposed this synthesis route in order to achieve a bio-based UPR with improved mechanical properties for composite applications. They investigated the mechanical and thermomechanical properties of completely cured bio-based UPR based on soybean oil, linseed oil and canola oil and presented their work in several scientific papers [4,5,6,7,8].
The crosslinking, or curing, of an UPR is characterized by a free radical polymerization mechanism. For this purpose, UPR is normally mixed with a reactive diluent, an initiator, an accelerator or promoter, and an inhibitor. One of the most widely used reactive diluent is styrene, because it has the feature to decrease the viscosity of the UPR and to increase the network density of the completely cured resin. Due to environmental and health issues caused by the high volatility of styrene, several works dealt with the development of harmless reactive diluents. These works propose that vinyl toluene, divinylbenzene, or acrylic acid derivatives are in fact less volatile alternatives that can reduce the health impact but not the environmental impact [9]. Other scientist have focused on the environmental aspect and developed reactive diluents based on various renewable resources [10,11,12]. Despite all attempts, a suitable bio-based substitute for styrene has still not been identified [3].
The thermal cure at room temperature or slightly increased temperature makes it necessary to choose a suitable combination of initiator and accelerator that generate free radicals at low processing temperature. An organic peroxide such as methyl ethyl ketone peroxide (MEKP) is a suitable initiator that decomposes rapidly in the presence of certain metallic carboxylate salts such as cobalt octoate [9]. Due to increasing health and environmental pressure on cobalt, the industry developed new harmless accelerators that are sufficiently effective to substitute cobalt-based accelerators [13]. Inhibitors are added to the mixture to prevent premature curing and to delay the beginning of the UPR polymerization, respectively. An effective inhibitor, for instance, is hydroquinone. The inhibitor has the responsibility to neutralize all the generated free radicals for a certain time. As soon as the inhibitor is consumed, the so-called induction period goes to completion. The free radical polymerization starts immediately and proceeds until the cure is complete [9].
Over the last three decades, the thermal curing kinetics of UPR system has been investigated by many researchers [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. Some of the studies determined the cure kinetic parameters as function of cure conversion by using ICKA methods [19, 26, 27, 33,34,35,36, 38]. Typical for ICKA methods is that the assumption of the cure mechanism or reaction model, f(α), is not required and that accurate ICKA methods are universally applicable to model the cure kinetics of simple single-step and complicated multi-step reactions as well [41,42,43,44]. Cure kinetics analysis can have either a practical or a theoretical purpose. The major practical purpose is the use of thermal cure kinetic parameters in the prediction of the progress of cure conversion, α, at arbitrary temperatures. In fact, obtaining the apparent Ea as a function of α by an accurate ICKA method is sufficient to predict the α(t)-curve at an arbitrary processing temperature [41,42,43,44]. However, the apparent pre-exponential factor, A, as a function of α as well as the reaction model, f(α) or g(α), can also be determined by using ICKA methods with the apparent Ea(α) [43, 44]. The whole set of apparent kinetic parameters can be further used to predict the α(t)-curve at an arbitrary processing temperature [45]. The induction period of an UPR system is not part of the crosslinking process. Therefore, another set of thermal cure kinetic parameters, which describe the effect of temperature on the induction period, were introduced [21].
Another typical transition of a thermosetting material during thermal cure, the chemical gelation, can be determined by using rheological measurements. A distinction may be drawn between molecular gelation and its result, the macroscopic gelation. Molecular gelation occurs at a well-defined conversion of the chemical reaction. It is dependent on the functionality, reactivity, and stoichiometry of the reactants and may be detected at the point where the loss tangent in dynamic rheology measurements becomes frequency independent. Macroscopic gelation, on the other hand, means estimation of molecular gelation and is evaluated at the G′–G′′ crossover point in a dynamic rheology measurement [46]. It is notable that the degree of cure conversion at the molecular gel point, αgel, is constant for a given thermosetting materials and independent of the cure temperature, i.e., gelation is isoconversional [47, 48].
The papers concerning bio-based UPR resin mixtures in the literature [4,5,6,7,8] were focused on the investigation of properties of completely cured resin mixtures, but not on the thermal cure kinetics. In the current work, for the first time, the thermal curing process of MaAELO resin mixture was studied systematically by using isothermal DSC measurements and dynamic rheology.
Theory of thermal cure kinetics based on isothermal experiments
Determination of α(t) by using DSC measurements
DSC is an effective method to monitor the thermal cure or crosslinking [49]. From each DSC experiment, the degree of cure in the course of time, α(t), can be calculated as the ratio of partly integrated cure enthalpy up to given time, H(t) (in J g−1), to the total cure enthalpy, Htotal (in J g−1):
To determine the total cure enthalpy, Htotal, it is recommended to measure the sample twice. In the first DSC run, the cure enthalpy H1 is determined, and in a second non-isothermal DSC run at a specific heating rate, each sample is measured again to determine the residual cure enthalpy, Hres. The total cure enthalpy, Htotal, is the sum of both enthalpies according to
Fundamental equations to analyze thermal cure kinetics
Chemical reactions including the curing reaction can be described by the rate of reaction dα/dt, which is directly proportional to the change in the concentration of chemical species, the rate constant k, and the reaction model, f(α) [49]:
The rate constant, k, shows a dependence on the reaction temperature. In 1889, Svante Arrhenius established an empirical relation for the temperature dependence of the reaction rate k(T):
A is the pre-exponential factor or Arrhenius frequency factor (in s−1), Ea is the activation energy (in J mol−1), R is the universal gas constant (8.314 J mol−1 K−1), and T is the absolute temperature (in K).
As a consequence, the temperature dependence of the reaction rate is based on the combination of Eqs. 3 and 4 [49]:
In principle, the thermal cure kinetics can be predicted for any temperature if the three kinetic parameters are known—the activation energy Ea, the pre-exponential factor A, and the reaction model, f(α).
Determination of Ea(α) by using ICKA methods
Classical kinetic analysis requires a priori knowledge on the reaction mechanism, f(α), and hence it is model-dependent. However, the crosslinking of thermosetting resins including the investigated resin mixture is a complex multi-step process rather than a simple chemical reaction of quantitatively known mechanism. Hence, the classical model-dependent kinetic analysis is not suitable to model the thermal cure kinetics because the exact reaction model is unknown. For complex processes, model-free kinetics or so-called isoconversional methods (ICKA) have been developed [41, 42] which assume that the Ea at a particular degree of cure, α, is independent of the temperature program (isoconversional principle).
The ICKA method of Vyazovkin (VA)
In the current work, the integral method of Vyazovkin in its advanced form (VA) [50] was used to determine Ea(α). All integral methods are based on the isoconversional principle implying that the integral form of reaction model, g(α) =\(\mathop \int \nolimits_{0}^{\alpha } \frac{{{\text{d}}\alpha }}{f(\alpha )}\), is constant at a given extent of α. In the case of isothermal DSC measurement, g(α) can be calculated directly by integrating Eq. 5. With α(t) from n = 3 isothermal experiments for a given extent of α, the integration resulted in
where the subscripts indicate the n = 3 different isotherms.
By dividing A, the pre-exponential factor is mathematically eliminated in Eq. 6. This forms up equations, one for each isotherm, in which Ea is the only unknown kinetic parameter.
For a given extent of α each combination of pairs of g(α) can be equated and divided by each other which gives n (n − 1) single equations or six single equations when n = 3:
All the n (n − 1) single equations can be totaled to form a new sum equation:
Because the VA approach is generalized to a minimization problem, it can be used for non-isothermal and isothermal DSC measurements as well, for isothermal DSC according to Eq. 9.
where the subscript n indicates different isotherms and Δα indicates the small interval of conversion. The time, t, and temperature, T, are the data of at least three cure conversion curves, α(t), and the Ea is the single unknown kinetic parameter.
In the case of variable Ea, it is important to solve Eq. 9 in small intervals of conversion, Δα, and in small time segments, Δt = (tα − tα−Δα), [42, 43]. The reason is that Ea can be rather highly variable over the whole range from 0 to α, but the actual value of Ea in a small range of Δα can be assumed to be constant as long as the interval is kept small enough. The Ea for a given interval of Δα is calculated by inserting the respective time and temperature from conversion curves of three or more different isothermal experiments into Eq. 9 first, and afterward by repeated variation of estimated values of Ea with the purpose to find a value of Ea for which the Ω-equation is a minimum. The existence of a function minimum is related to the fact that Ω has the shape of a parabola that is opening toward the top and the vertex is placed at the y-coordinate n (n − 1). The procedure of minimization has to be repeated for different extents of α to achieve further values of Ea in the α-range between 0 and 1. In the case of a chemical single-step process, the apparent Ea of each α is constant, but sometimes the values of Ea will show dependence on α, which reveals a complex multi-step process.
The ICKA method of Friedman (FR)
To get the well-known equation of a straight line, y = k x + d, the present work used Flynn’s adaptation of the Friedman method (FR) [51], which has no limitation on the temperature program [52]:
where \(y = T \ln \left( {\frac{{{\text{d}}\alpha }}{{{\text{d}}t}}} \right)\), \(x = T\) (in K), \(k = \left[ {\ln A f(\alpha )} \right]\), and \(d = - \frac{{E_{\text{a}} }}{R}\).
After linear regression of the plot of pair of values \(y = T \ln \left( {\frac{{{\text{d}}\alpha }}{{{\text{d}}t}}} \right)\) and x = T for a given α from at least three cure conversion curves, α(t), the apparent Ea at the given conversion can be determined from the intercept, d, of the regression line. Furthermore, the slope, k, of the linear regression holds the apparent A and the reaction model, f, at the given conversion. This value can be used in the reconstruction of f(a) as soon as A was determined by an appropriate method (see next section). By repeating the linear regression for different extents of α, further values of Ea and \(\left[ {\ln A f(\alpha )} \right]\) in the α-range between 0 and 1 are determined. A more or less constant Ea in the course of α normally indicates a single-step process, whereas variations are a signal of a multi-step process.
Determination of A(a) by using the compensation effect (KCE)
First, it is important to note that the kinetic parameters Ea and A in terms of transition state theory [53] are different from Ea and A resulting from mathematical fitting methods like ICKA methods [54]. The former parameters are not correlated and have their specific physical meaning, Ea links to an energy barrier and A to the vibrational frequency of the activated complex. On the other hand, the parameters derived from experimental data using ICKA methods are mathematical fitting values, which are correlated. Therefore, it makes independent interpretation of both values impossible. This effect is known as the so-called compensation effect (KCE) [55]. In order to emphasize the differences, Ea and A calculated by ICKA methods are called “apparent” activation energy and “apparent” pre-exponential factor, respectively. If the reaction is the sum of multi-step reactions, the ICKA methods produces values of apparent Ea and A that may vary in the course of α. In that case, the apparent values are “overall” values that reflect the overall reaction process [43].
Due to the correlation effect, it is possible to take the apparent Ea(α) to determine the correlated apparent A(α) [42, 44]. In the case of isothermal experiments, three consecutive linear regressions based on Eqs. 11, 12, and 13 are used to determine the A(α). The starting point of the calculation is the integral form of the reaction model:
where the subscript i indicates a constant isothermal temperature.
To solve Eq. 11, any f(α) or its integral form, g(α), has to be assumed by picking one from a list of theoretical reaction models published in the literature [42, 44]. With the assumed mathematical expression of g(α), the rate constant k(Ti) at a given isothermal temperature, Ti, is calculated from the slope of the linear regression of the plot of g(α) at various α versus the time t to reach the respective α. The time values, tα, are taken from one α(t)-curve derived from the respective isothermal DSC experiment. This procedure is repeated for at least i = 3 isothermal DSC experiments to achieve the rate constant k(Ti) of each isothermal temperature.
With the rate constants at different isotherms, k(Ti), Eq. 4 is transformed into:
From the plot of k(Ti) against the inverse of temperature, 1/Ti, of at least three isothermal temperatures, a single value of Ea is calculated from the slope, and another single value of ln A is calculated from the intercept of another linear regression. Another pair of correlated values can be calculated by repeating the calculation of Eqs. 11 and 12 with another assumption of g(α). Each pair of values are numbers only if an inaccurate reaction model is chosen but due to the compensation effect, the pair of values based on different reaction models, Ej and Aj, must form a straight line on which also E and A values based on the accurate reaction model lie according to
where the subscript j indicates the pair of values of each assumed reaction model.
The slope b and the intercept a of this third linear regression line are called compensation parameters.
Finally, the Ea(α), which is determined by using the VA or FR method before, can be inserted together with compensation parameters into Eq. 13 to determine the correlated apparent A(α).
The reconstruction of f(α)
After the determination of both apparent kinetic parameters, Ea(α) and A(α), it is possible to reconstruct the f(α). In the case of Ea(α) calculated with the VA method, Ea(α) and the corresponding A(α) can be inserted together with the reaction rate dα/dt from any isothermal DSC experiment in Eq. 5 to reconstruct f(α). In the case of FR method, values of ln(A(α)) can be directly inserted into the slope ln[A·f(α)] which is an additional result of linear regression to determine Ea(α) from the intercept (see Eq. 10).
Prediction of thermal cure kinetics by using apparent Ea(α) only
The starting point to predict α(t)-curves at arbitrary isothermal temperatures on the basis of only the apparent Ea(α) is a slight adaption of Eq. 6 according to:
The subscript 1 on the left-hand side of Eq. 14 indicates values of g(α) taken from one of the thermal measurements at any isotherms used for the computation of Ea(α), whereas the subscript 2 on the right-hand side indicates values of the same g(α) at another isothermal temperature, for which no measurement is available. After dividing A on both sides of Eq. 14 and rearrangement, the time, t2, to reach a given extent of α at the arbitrary isothermal temperature, T2, can be predicted according to
Considering the variable Ea, the left-hand side of Eq. 14 can be modified in order to take small intervals of conversion, Δα, and small time segments, Δt = (tα − tα−Δα), respectively, into account:
The total time prediction, tα, for an arbitrary isothermal temperature, T2, to reach a given extent of α can be calculated by summing up all time intervals, tΔα, predicted in each iteration step, i, from α0 = 0 up to the given α and by taking the actual Ea of time interval or iteration step i, respectively, into account according to:
Prediction of thermal cure kinetics by using the whole set of all three kinetic parameters
Another kinetic prediction equation was developed based on Eq. 5 using the whole set of all three kinetic parameters [45]. After rearrangement of Eq. 5, the integration leads to the equation
where T2 is the isothermal temperature, for which no measurement is available.
Equation 18 is to be adapted to get an accurate time prediction of small time segments when apparent kinetic parameters are variable in the course of α but allowed to be assumed as constant values in every small time segment, Δt = (tα − tα-Δα):
T2 in Eq. 19 is the isothermal temperature, for which no measurement is available.
The total time prediction, tα, for an arbitrary isothermal temperature, T2, to reach a given extent of α can be calculated again by using Eq. 17.
Validation of kinetic parameters determined by ICKA methods
The equations for prediction (Eqs. 16 or 19 and 17) provide an opportunity to validate the accuracy of determined apparent kinetic parameters and the accuracy of chosen ICKA method, respectively, by comparison of predicted α(t)-curves and α(t)-curves from thermal experiments. Ideally, the validation encompasses not only the comparison with α(t)-curves from the thermal experiments used in the determination of kinetic parameters but includes also α(t)-curves from thermal experiments at other temperatures.
Experimental
Materials
Cold pressed linseed oil was purchased from Makana Produktion und Vertrieb GmbH, Germany. Glacial acetic acid was supplied by VWR International, Austria. The 35% solution of hydrogen peroxide was purchased from Merck, Germany. The suppliers of Purolite C150H and sodium chloride were Purolite GmbH, Germany, and Carl Roth, Germany, respectively. Triphenylphosphine, hydroquinone, acrylic acid, maleic anhydride, and styrene were purchased from Sigma-Aldrich. Methyl ethyl ketone peroxide FL 505 S was obtained from Vosschemie, Germany, and the manganese-based accelerator DriCAT 2700F was obtained by Dura Europe, Spain.
Synthesis of thermosetting resin from linseed oil
Cold pressed linseed oil was epoxidized (ELO) through in situ epoxidation in the presence of glacial acetic acid and hydrogen peroxide. In a round-bottomed flask, 100 g of linseed oil was mixed with 14.87 g of glacial acetic acid along with 10 g of Purolite ion exchange catalyst. 111 g of hydrogen peroxide was weighed accurately and added to the reaction mixture in two portions. The temperature of the medium was kept constant at 75 °C for 9 h under constant stirring. At the end, purolite was filtered off and neutralized using sodium bicarbonate in a separating funnel. Saturated sodium chloride solution was used to separate water and oil mixture. The excess water was removed under vacuum and dried using sodium sulfate. The epoxy equivalent (EEW) of the ELO was calculated using titration method.
Afterward, an acrylated epoxidized linseed oil (AELO) was synthesized in the laboratory using the following procedure. Initially, the mixture of ELO (53 g) and triphenylphosphine (3.19 g) containing 0.06 mass% hydroquinone (with respect to ELO) was stirred at a temperature of 70 °C for 20 min. Acrylic acid (15.44 g) was added to the mixture drop by drop for about 20 min, and the mixture was stirred for another 6 h at 70 °C. The effect of acrylation process was checked by using an infrared (IR) spectrometer (Bruker Equinox 55) with an attenuated total reflectance (ATR) unit.
Without any purification, the AELO was further maleinated in the presence of maleic anhydride. 50 g of AELO was reacted with 4 g of maleic anhydride at the temperature of 70 °C for 40 min. After that, the reaction mixture was cooled down to 40 °C. The final maleinated AELO (MaAELO) was stored below 16 °C for further characterization.
The synthesis route for obtaining renewable thermosetting resin from vegetable oil is shown in Fig. 1. The epoxidation is carried out by converting the double bonds of unsaturated fatty acids in vegetable oil into oxirane rings, which are further converted into acrylic groups during acrylation. Further, in the maleination process, the hydroxyl groups in the acrylic product react with maleic anhydride that gives acid end groups and adds more unsaturation to the final product.

Scheme of the synthesis route to achieve highly reactive thermosetting resin from vegetable oil
Rheological measurements to determine the effect of reactive diluent
MaAELO was mixed with increased amounts of styrene as reactive diluent (20, 25, 30 mass%). The shear rate dependency of viscosity of each resin mixture was measured with a Physica MCR 101 (Anton Paar GmbH, Graz, Austria) for two isothermal temperatures, 25 °C and 60 °C, in order to find the minimum amount of styrene which is needed to achieve a suitable low viscosity for the processing with liquid composite molding (LCM) techniques.
Preparation of resin mixture
The MaAELO resin was mixed with styrene in the optimum proportions found by rheological measurements. Then, 2 mass% of methyl ethyl ketone peroxide (MEKP) and 1 mass% of manganese-based accelerator DriCAT 2700F (Dura Europe, Spain) were added to achieve the final MaAELO resin mixture.
DSC measurements and analysis of thermal cure
The MaAELO resin mixture was measured isothermally (40, 45, 50, 55, and 60 °C) in a first DSC run with a Mettler Toledo (Greifensee, Switzerland) 822e DSC to determine the cure enthalpy H1. In a second DSC run, each sample was measured again at 5 °C min−1 to determine the residual cure enthalpy, Hres. The total cure enthalpy, Htotal, was the sum of both enthalpies. All DSC measurements were performed under inert atmosphere (N2).
The thermal data and α(t)-curves derived from the three isothermal DSC experiments at 40, 50, and 60 °C were used to determine the whole set of all three apparent cure kinetic parameters of MaAELO resin mixture according to the equations in the theory section. The reconstruction of f(α) on the basis of Ea(α) determined by VA method and the respective A(α) by using Eq. 5 was performed together with the reaction rate dα/dt taken from the isothermal DSC experiment at 50 °C.
The validation of the apparent kinetic parameters included the comparison of predicted α(t)-curves with α(t)-curves from all five isothermal experiments at 40, 45, 50, 55, and 60 °C.
Modeling and prediction of the induction period
Isothermal DSC measurements are helpful to check whether the cross-linking of an UPR system starts immediately at a given isothermal temperature, or the cross-linking is retarded and starts later after an induction period, tind. In the latter case, the time when the DSC curve begins to deviate from the baseline can be related to tind. Under the assumption that tind follows the Arrhenius relation, the temperature dependence of tind can be described as follows [21]:
Eind is the activation energy (in J mol−1), tind_0 is a time factor (in s), R is the universal gas constant (8.314 J mol−1 K−1), and T is the absolute temperature (in K).
The parameters Eind and tind_0 are the characteristic kinetic parameters of an UPR system to model its induction period at arbitrary temperatures. To determine both parameters of the MaAELO resin mixture, the induction periods, tind (in min), of the five isothermal (40, 45, 50, 55, and 60 °C) DSC experiments were taken and plotted as a function of temperature. From the plot of the logarithm of measured induction period, ln(tind), versus the respective inverse temperature (1000/T), it was possible to calculate the single value of Eind (in kJ mol−1) from the slope and the time factor, tind_0 (in min), from the intercept of linear regression.
Both kinetic parameters were used to predict the induction period of MaAELO resin mixture for different isothermal temperatures and to compare the predicted times with the induction period of isothermal DSC experiments.
Rheological measurements and analysis of thermal cure
Dynamic rheological measurements in oscillatory mode and using a temperature program provide the monitoring of the complex progress of viscoelastic properties during thermal cure. Three parameters, the complex viscosity, η*, the storage modulus, G′, and the loss modulus, G′′, are recorded versus time and temperature. The actual G′ is related to the actual elastic state and the actual G′′ to the actual viscous state of the reactive mixture in the progress of cross-linking. The ratio of G′′ to G′ is defined as the loss tangent, tan δ, according to
The thermal cure of MaAELO resin mixture was analyzed by dynamic rheology with a Physica MCR 101 (Anton Paar GmbH, Graz, Austria) between parallel plates in dynamic oscillatory mode with simultaneous multiwave frequency sweeps (deformation = 3%; frequencies = 5, 10, 20, and 30 s−1) at 45 °C and again at 50 °C. Because at the molecular gel point, tan δ has to be independent of the frequency, it was able to detect the gel point of both isothermal temperatures at the time when the tan δ-curves of the simultaneous multiwave frequency sweeps passed through a single point [56]. Afterward, the measurement was continued at an isothermal temperature until each frequency reached its G′–G′′ crossover point where tan δ = 1. Then, the sample pan containing the mixture was immediately transferred into a bath of liquid nitrogen to preserve the degree of cure conversion when the measurements were stopped by cooling down the mixture as quickly as possible. The partially cured samples were measured again by DSC at 5 °C min−1 to determine the degree of cure at the molecular gel point, αgel, and at the respective crossover points.
Measurements to determine the Tg of cured mixtures
Samples of cured MaAELO resin mixture (cured at 25 °C for 72 h without post-curing and inclusive post-curing at 80 °C for 8 h) were measured at 10 °C min−1 with a Mettler Toledo (Greifensee, Switzerland) TMA/SDTA 840 to determine the Tg of the crosslinked resin mixture according to the standard ASTM E1545 [57]. In this context, the Tg was determined from the point of intersection of the tangents to the TMA curve (onset temperature). In addition, dynamic DSC experiments were performed three times in a row (temperature range: 25–150 °C; heating rate: + 10 °C min−1; cooling rate: − 10 °C min−1). The DSC curve of the last heating program was used to determine the Tg of the fully cured MaAELO resin mixture.
Results and discussion
Characterization of synthesis products
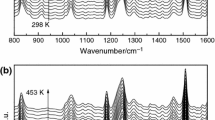
In the first synthesis step, the double bonds of unsaturated fatty acids in linseed oil (LO) were transformed into oxirane rings forming an epoxidized linseed oil (ELO). The average molecular weight of the linseed oil after epoxidation was 1020–1040 g mol−1, and the content of double bond was 5.4 per triglyceride molecule. The epoxy equivalent (EEW) of the final ELO determined by using titration method was in the range between 180 and 190. In the next synthesis step, the oxirane was converted into acrylic groups forming an acrylated epoxidized linseed oil (AELO). FT-IR measurements reveal that 80–85% of the oxirane was converted into acrylate groups. Some of oxirane remained just after acrylation, which could be evidenced by the remaining signal of oxirane groups at 830–850 cm−1 (Fig. 2).

IR spectrum of LO (a), ELO (b), and AELO (c)
Rheological measurements and preparation of resin mixture
Like conventional petrochemical UP resins, the pure MaAELO resin also had a viscosity, which was much too high for use in the processing of fiber-reinforced composites by liquid composite molding (LCM) techniques. Therefore, MaAELO was mixed with increased amounts of styrene as reactive diluent (20, 25, 30 mass%). The viscosity of each resin mixture was measured at two isothermal temperatures, 25 °C and 60 °C. By adding 30 mass% styrene, the mixture achieved firstly a viscosity of about 1250 mPa s at 25 °C and nearly 200 mPa s at 60 °C, respectively, which was sufficiently low for LCM techniques. Based on the chosen MaAELO/styrene mixture, the resin preparation was finished by adding 2 mass% of MEKP and 1 mass% of the cobalt-free accelerator DriCat 2700F to the mixture. It should be noted that no inhibitor was added to the resin mixture, but in fact, an inhibitor was present because hydroquinone was used in the synthesis of MaAELO.
DSC measurements
The thermal cure of MaAELO resin mixture was analyzed by using DSC. Five isothermal experiments (40, 45, 50, 55, and 60 °C) were performed in a first DSC run (Fig. 3), followed by a second DSC run at a heating rate of 5 °C min−1 to check for a residual cure enthalpy.

DSC heat flow of MaAELO resin mixture at isotherms (a) 40, (b) 45, (c) 50, (d) 55, and (e) 60 °C including vertical dashed lines to mark the induction periods of each isotherm
The resin mixture reached its characteristic total cure enthalpy at about 280 J g−1. At isotherms of 50 °C or more, the first enthalpy reached always the total cure enthalpy. At lower temperatures, the residual cure enthalpy was about 10 J g−1. This indicated the effect of vitrification at low temperatures, although the effect was only small and so the resin mixture reached at least 97% of cure conversion in the first DSC run. The isothermal DSC experiments revealed also the effect of hydroquinone. Hence, the cure started not immediately but after a remarkable induction period tind (vertical dashed lines in Fig. 3). With the progress of enthalpy after the induction period, the cure conversion curves, α(t), were calculated for each isotherm which was the basis for deriving the thermal cure kinetic parameters.
Modeling and prediction of induction period
After plotting the logarithm of measured induction period, tind, versus the inverse isothermal temperature, a single value of Eind was calculated from the slope (= 64.4 kJ mol−1; confidence interval: lower 95% = 43.8, upper 95% = 85.0) and a time factor, tind_0, from the intercept (= 2.2*10−7 s; confidence interval: lower 95% = 1*10−10, upper 95% = 4.7*10−4) of linear regression (r2 = 0.97; standard error of estimate (SEE) = 0.02 min) (Fig. 4).

Linear regression of measured induction period, tind, and inverse of isothermal temperature from DSC experiments
Both kinetic parameters were used to predict the induction period for different isothermal temperatures (Table 1).
ICKA methods to determine apparent Ea(α)
Based on the conversion curves from three isothermal DSC experiments (40, 50, and 60 °C), the apparent Ea(α) was calculated by using two ICKA methods, the integral method of Vyazovkin (VA) and the differential method of Friedman (FR) (Fig. 5).

Apparent Ea(α) for thermal cure of MaAELO resin mixture calculated with ICKA methods of VA and FR
Both methods resulted in similar Ea(α)-curves. The apparent activation energy of both methods remained nearly constant at 25 kJ mol−1 until cure conversion reached 50%, and increased afterward up to nearly 60 kJ mol−1 in the final stage of cure. Otherwise, the FR method resulted in low coefficient of determination, r2, especially in the beginning of cure, while the VA method started to deviate significantly from optimum only at α higher than 90% (Table 2). The variable dependency of Ea on the curing degree, α, reveals that curing reaction is as a complex multi-step process. The increase in Ea at α beyond 50% can be related to diffusion effects due to increase in viscosity when α increases.
Compensation effect to determine A(α)
Table 3 shows the pairs of correlated kinetic parameters Ea and ln(A) obtained from two consecutive linear regressions based on Eqs. 11 and 12 by using the conversion curves of three isothermal DSC experiments (40, 50, and 60 °C) and five different reaction models taken from the literature [42, 44].
Based on the pairs of correlated kinetic parameters, the compensation parameters were calculated from the linear regression according to Eq. 13 (Fig. 6). The obtained values from linear regression were b = 0.353 mol kJ−1 (confidence interval: lower 95% = 0.270, upper 95% = 0.435), a = −3.165 min−1 (confidence interval: lower 95% = −5.706, upper 95% = −0.624), standard error of estimate (SEE) = 1.54 min−1, and r2 = 0.984.

Plot of ln A and Ea for five different reaction models—Mampel first order (1), Avrami–Erofeev A2 (2), Contracting cylinder R2 (3), One-dimensional diffusion D1 (4), and Three-dimensional diffusion D3 (5)—to determine the compensation parameters by linear regression
With the compensation parameters, the A(α)-curve could be calculated by inserting the values of apparent Ea(α)-curves, which were already determined with both the VA and FR method, into Eq. 13 (Fig. 7 and Table 4). Due to the correlation of apparent Ea and apparent A, the curve of A(α) was similar to the curve of Ea(α). The A of both methods kept nearly constant (AVA ≈ 251 min−1 and AFR ≈ 270 min−1) until cure conversion reached 50%, and increased afterward up to a maximum (AVA = 3.93 × 107 min−1 and AFR = 1.10 × 108 min−1) in the final stage of cure.

Apparent A(α) for thermal cure of MaAELO resin mixture obtained using the compensation parameters and the apparent Ea(α) based on VA method and FR method
Reconstruction of f(α)
After determination of Ea(α) and A(α) with both ICKA methods, it was possible to reconstruct the last kinetic parameter for the MaAELO resin mixture, the f(α) (Fig. 8). In case of Ea(α) calculated with the VA method, Ea(α) and corresponding A(α) were inserted together with the reaction rate dα/dt from isothermal DSC experiment at 50 °C into Eq. 5 to determine f(α). In case of FR method, the slope of linear regression to determine Ea(α) (Table 2) could be used to calculate f(α) with the corresponding A(α). All data needed to determine f(α) and the calculation of f(α) for both methods are summarized in Table 4.

Reconstruction of f(α) for thermal cure of MaAELO resin mixture based on VA and FR kinetic parameters in comparison with reaction model of Avrami–Erofeev with n = 2.5
The values of reconstructed f showed a similar dependency on the curing degree, α, as the reaction rate dα/dt of any isotherm (see Table 4). The values of f and reaction rate were low in the beginning of cure and grew up to a maximum in the α-range of 40–50%. Afterward, the values dropped and ended at values much lower than in the beginning of cure. The shape of f(α) and dα/dt corresponded to an autocatalytic behavior where f(α) and the rate passes through a maximum. For the reaction of UPR, mostly the phenomenological/empirical model of Kamal [58] or Sourour-Kamal [59] have been used in order to model the cure behavior of UPR that undergo a combination of nth-reactions and autocatalytic reactions during thermal cure. Avrami–Erofeev models are other mathematical equations that are able to describe autocatalytic reactions [25]. In the current work, the reconstructed f based on VA parameters showed a very good correlation with the Avrami–Erofeev reaction type when the factor n = 2.5 (Fig. 8). This does not mean that Avrami–Erofeev is the true underlying reaction model of the MaAELO resin mixture, but it shows definitely the best mathematical fit. The reason for this uncertainty is the ambiguity of reaction models and so the mathematical fit may be successful despite the fact that an obviously wrong reaction model was used [60].
Comparison of prediction of thermal cure by using only apparent Ea(α) with the prediction using the whole set of all three kinetic parameters
The curves of apparent Ea(α) enabled the prediction of α(t)-curves at arbitrary temperatures without knowledge of the other two kinetic parameters, the pre-exponential factor, and the reaction model. The α(t)-curves were predicted using Ea(α) from the VA method according to Eq. 16 followed by Eq. 17 and again with Ea(α) from the FR method. The predicted α(t)-curves were compared with α(t)-curves calculated from isothermal DSC measurements (40, 45, 50, 55, and 60 °C) (VA results in Fig. 9 and FR results in Fig. 10).

α(t)-curves obtained from isothermal DSC experiments (-X-) at five isotherms (a) 40, (b) 45, (c) 50, (d) 55, and (e) 60 °C compared with α(t)-curves predicted using Ea(α) from VA method at the same isotherms (open circle) 40, (open diamond) 45, (filled square) 50, (filled triangle) 55, and (filled circle) 60 °C

α(t)-curves obtained from isothermal DSC experiments (-X-) at five isotherms (a) 40, (b) 45, (c) 50, (d) 55, and (e) 60 °C compared with α(t)-curves predicted using Ea(α) from FR method at the same isotherms (open circle) 40, (open diamond) 45, (filled square) 50, (filled triangle) 55, and (filled circle) 60 °C
The prediction based on the single apparent kinetic parameter Ea(α) was similar regardless of whether Ea(α) from VA method (Fig. 9) or FR method (Fig. 10) was used. The prediction at 50, 55, and 60 °C was very precise, while at 40 °C and 45 °C, when the thermal cure was affected by vitrification, the prediction differed from the experimental curves. However, the deviations of vitrified system were marginal. Therefore, it could be concluded that VA method and FR method as well were valid to analyze the thermal cure process of the bio-based MaAELO resin mixture. With both methods, the progress of apparent Ea as function of α could be determined accurately, and this single kinetic parameter was sufficient to predict precisely the thermal cure at arbitrary isothermal process temperatures. For 40 °C and 45 °C, the predicted time to reach a specific degree of cure was always earlier than the measured time. Interestingly, the prediction deviations for both temperatures started far below of the point of vitrification, which was determined at 97% of conversion. Obviously, the vitrification point was not the only factor that was responsible for the change from the chemically controlled rate to the diffusion-controlled rate. A comparable early start of diffusion-controlled curing rate was presented in the paper of Sbirrazzuoli et al. [61]. Sbirrazzuoli and co-workers figured out that the curing rate already changed to the diffusion-controlled rate when the gel point occurred at a low degree of cure. Thus, at this point, we propose the hypothesis that gelation of MaAELO resin mixture was responsible for the early beginning of deviations in the prediction. Testing the hypothesis is included in the next sections when the rheological properties of the MaAELO resin mixture are discussed. Furthermore, the prediction for 40 °C was slightly more accurate than the prediction for 45 °C. This can be explained by the fact that the conversion curve at 40 °C was one of three conversion curves to derive the model parameter, Ea(α). Hence, the higher accuracy of prediction for 40 °C was not more than a side effect of the modeling procedure.
Another prediction was performed by using the whole set of kinetic triplet calculated with the VA method (Ea_VA(α), AVA(α), and fVA(α)) and FR method (Ea_FR(α), AFR(α), and fFR(α)) according to Eq. 19 followed by Eq. 17. The comparison of the prediction with α(t)-curves calculated from isothermal DSC measurements (40, 45, 50, 55, and 60 °C) is shown in Fig. 11 (VA) and Fig. 12 (FR).

α(t)-curves obtained from isothermal DSC experiments (-X-) at five isotherms (a) 40, (b) 45, (c) 50, (d) 55, and (e) 60 °C compared with α(t)-curves predicted using all three kinetic parameters Ea_VA(α), AVA(α), and fVA(α) at the same isotherms (open circle) 40, (open diamond) 45, (filled square) 50, (filled triangle) 55, and (filled circle) 60 °C

α(t)-curves obtained from isothermal DSC experiments (-X-) at five isotherms (a) 40, (b) 45, (c) 50, (d) 55, and (e) 60 °C compared with α(t)-curves predicted using all three kinetic parameters Ea_FR(α), AFR(α), and fFR(α) at the same isotherms (open circle) 40, (open diamond) 45, (filled square) 50, (filled triangle) 55, and (filled circle) 60 °C
While the prediction in a model-free way by using Ea_VA(α) (Fig. 9) or Ea_FR(α) (Fig. 10) was very precise, the prediction using the apparent kinetic triplet from VA method (Fig. 11) or FR method (Fig. 12) failed. The failed predictions started to differ already in the early stages of cure conversion. Considering the results of FR method in Table 2, it is obvious that the coefficient of determination r2 of linear regression is very low in the beginning of thermal cure, but grows up to reasonable values in the later progress of cure. The low r2 in the beginning of cure could be explained by the assumption of imprecise rate of conversion derived from the isothermal DSC experiments in the beginning of cure, while the higher r2 indicated that the rates of conversion later were more accurate. The issue of erroneous rate of conversion could be completely avoided when Ea(α) was determined by using the VA method, and the prediction was based on Eq. 16 followed by Eq. 17 (Fig. 9), because none of the used equations was based on the rate of conversion. In case of VA method, at least in the determination of f(α), the rate of conversion was needed. Hence, the values of f(α) in the beginning of cure were erroneous, and the prediction using Eq. 19 followed by Eq. 17 with all kinetic parameters failed (Fig. 11). In case of FR method, already the linear regression to determine Ea(α) and [ln(A·f(α)] was based on the rate of conversion. Interestingly, the error was negligible when only the single kinetic parameter Ea(α) from the slope of linear regression was used to predict α(t)-curves using Eq. 16 followed by Eq. 17 (Fig. 10). Nonetheless, the prediction using Eq. 19 followed by Eq. 17 with all kinetic parameters from FR method failed (Fig. 12). Therefore, the imprecise rate of conversion in the beginning of cure was not the only reason why the prediction with all kinetic parameters failed, while the prediction with only Ea(α) was successful. Considering the strong correlation between the apparent kinetic parameters, there was another notable factor that affected the accuracy of predictions. The determination of A(α) depended strongly on the accuracy of compensation parameters, and A(α) was important for the reconstruction of f(α). So, the advantage of kinetic calculations based on Ea(α) alone was, to avoid the errors in determination of the whole set of apparent kinetic parameters and, instead, to rely on the accurate determination of Ea(α), which was absolutely sufficient to predict the thermal cure of the complicated curing process of MaAELO resin mixture.
Rheological measurements and analysis of thermal cure
The MaAELO resin mixture was measured with a rheometer in dynamic oscillatory mode and with simultaneous multiwave frequency sweeps at two isothermal curing temperatures. For 45 °C, the tan δ of MaAELO resin mixture as function of time is shown in Fig. 13 and for 50 °C in Fig. 14.

tan δ of MaAELO resin mixture as function of time obtained from dynamic rheology at 45 °C and for the frequencies (filled diamond) 5, (filled triangle) 10, (filled square) 20, and (filled circle) 30 s−1 including vertical dashed line to mark the molecular gel point

tan δ of MaAELO resin mixture as function of time obtained from dynamic rheology at 50 °C and for the frequencies (filled diamond) 5, (filled triangle) 10, (filled square) 20, and (filled circle) 30 s−1 including vertical dashed line to mark the molecular gel point
The time at which the tan δ became frequency independent has been marked as the molecular gel point (after 7071 s of thermal curing at 45 °C and after 4215 s at 50 °C). The partially cured samples of the interrupted rheological measurements were measured by dynamic DSC. The DSC measurements confirmed the assumption of isoconversional behavior of the gel point. The gel point times at the two isotherms were different, but considering the thermal cure kinetics, these times related to the same degree of cure, αgel. The MaAELO resin mixture reached gelation already in a very early phase of curing (αgel < 5%). This result confirmed the aforementioned hypothesis that the early beginning of deviations of prediction is related to the early occurrence of gelation. The gel point occurred not at the angle of 45 degrees where the tan δ was 1 but at an angle of 81 degrees for 45 °C and 85 degrees at 50 °C.
The crossover points of G′ and G′′ occurred later than the gel point and the time of crossover points grew up with increasing frequency. The rise of crossover points with increasing frequency in comparison with the gel point is shown in Fig. 15 for the isothermal curing temperature of 45 °C and in Fig. 16 for the isothermal curing temperature of 50 °C.

Crossover points of G′ and G′′ as a function of frequency (filled square) for MaAELO resin mixture obtained from dynamic rheology at 45 °C in comparison with the molecular gel point (filled diamond)

Crossover points of G′ and G′′ as a function of frequency (filled square) for MaAELO resin mixture obtained from dynamic rheology at 50 °C in comparison with the molecular gel point (filled diamond)
With increasing frequencies, the G′–G′′ crossover points shifted to longer periods, but actually maximal 120 s after the gel point.
Measurements to determine the Tg of cured mixtures
Additionally to the thermal cure, the Tg, of the thermally cured MaAELO resin mixture was determined from TMA curves. The Tg of MaAELO resin mixture cured at 25 °C reached 43 °C and increased up to a maximum of 55 °C when the cured resin was post-cured at 80 °C. The Tg of fully cured resin mixture was determined from the last of three heating programs in dynamic DSC experiment. This Tg at 54 °C was in good agreement with the Tg determined from the TMA curve of the post-cured MaAELO resin.
The final Tg at 54 °C or 55 °C confirmed the effect of vitrification observed in the isothermal DSC measurements at 40 and 45 °C. For both curing temperatures, the rising Tg during curing process came close to the respective cure temperature and caused the disruption of any further rise of conversion. However, the degree of cure was already 97% when this point of vitrification occurred. As stated before, the observed deviation between predicted and measured conversion was not an effect of vitrification but rather an effect of gelation in the early stage of cure. At a curing temperature of 50 °C and higher, the resin system did not vitrify because the curing rate of the resin system was sufficiently high to reach the fully cured state before the Tg came close to the curing temperature.
Conclusions
In the current work, for the first time, the thermal curing behavior of maleinated acrylated epoxidized linseed oil (MaAELO) was studied systematically by using isothermal DSC and dynamic rheology. The systematic investigations encompassed (1) the determination of the whole set of apparent kinetic parameters, Ea(α), A(α), and f(α) by using two accurate ICKA methods (VA and FR) and the compensation effect, (2) the prediction of cure conversion curve, α(t), at arbitrary processing temperatures with all three kinetic parameters were compared with the prediction based on Ea(α) alone, and (3) the determination of molecular and macroscopic gelation during thermal cure of MaAELO resin mixture. The variable dependency of Ea(α) revealed that the curing reaction of the MaAELO resin mixture was a complex multi-step process. Ea(α) was nearly constant around 25 kJ mol−1 when α < 50%, and increased afterward up to 65 kJ mol−1. The prediction in a model-free way only using Ea(α) was very accurate. The accuracy of prediction was not improved but decreased when the other two kinetic parameters were taken into account. The MaAELO resin mixture reached gelation already in a very early phase of curing (αgel < 5%). This result confirmed the assumption that gelation was responsible for the change of the reaction to diffusion-controlled process even at the very early stage of cure. Another effect that affected the thermal cure at processing temperatures below 50 °C was the effect of vitrification. However, at 40 °C and 45 °C, the degree of cure has already reached 97% conversion when the point of vitrification occurred. At curing temperature of 50 °C and higher, the curing rate was sufficiently high to reach the fully cured state before vitrification. Due to vitrification, the Tg of MaAELO resin mixture cured at 25 °C reached a maximum of 43 °C and when the cured resin was post-cured at 80 °C the Tg increased up to 55 °C.
References
Raquez JM, Deléglise M, Lacrampe MF, Krawczak P. Thermosetting (bio)materials derived from renewable resources: a critical review. Prog Polym Sci. 2010;35:487–509.
Ma S, Li T, Liu X, Zhu J. Research progress on bio-based thermosetting resins. Polym Int. 2016;65:164–73.
Li Q, Ma S, Xu X, Zhu J. Bio-based unsaturated polyesters. In: Thomas S, Hosur M, Chirayil CJ, editors. Unsaturated polyester resins. fundamentals, design, fabrication, and applications. Amsterdam: Elsevier; 2019.
Lu J, Wool RP. Development of new SMC resins and nanocomposites from plant oils. In: Proceedings of the 4th annual SPE automotive composites conference, Troy; 2004.
Lu J, Khot S, Wool RP. New sheet molding compound resins from soybean oil I, Synthesis and characterization. Polym. 2005;46:71–80.
Lu J, Wool RP. Novel thermosetting resins for SMC applications from linseed oil: synthesis, characterization, and properties. J Appl Polym Sci. 2006;99:2481–8.
Lu J, Wool RP. Sheet molding compound resins from soybean oil: thickening behavior and mechanical properties. Polym Eng Sci. 2007;47:1469–79.
Fahimian M, Adhikari D, Raghavan J, Wool RP. Biocomposites from canola oil based resins and hemp and flax fibers. In: 13th European conference on composite materials 2008 Stockholm, Sweden. ECCM13; 2013.
Krämer H. Polyester resins, unsaturated. Ullmann’s encyclopedia of industrial chemistry, vol. 28. Weinheim: Wiley; 2012, p. 613–622.
Zhang Y, Li Y, Wang L, Gao Z, Kessler MR. Synthesis and characterization of methacrylated eugenol as a sustainable reactive diluent for a maleinated acrylated epoxidized soybean oil resin. ACS Sustain Chem Eng. 2017;5:8876–83.
Zhang Y, Li Y, Thakur VK, Wang L, Gu J, Gao Z, Fan B. Wu Q, Kessler MR. Bio-based reactive diluents as sustainable replacements for styrene in MAESO resin. RSC Adv. 2018;8:13780–13788.
Zhang Y, Thakur VK, Li Y, Garrison TF, Gao Z, Kessler MR. Soybean oil-based thermosetting resins with methacrylated vanillyl alcohol as bio-based reactive diluent. Macromol Mater Eng. 2018. https://doi.org/10.1002/mame.201700278.
Zuijderduin R, Böttger W. Cobalt-free curing taking off. Reinf Plast. 2013;57(1):29–32.
Cuadrado TR, Borrajo J, Williams RJJ, Clara FM. On the curing kinetics of unsaturated polyesters with styrene. J Appl Polym Sci. 1983;28(2):485–99.
Avella M, Martuscelli E, Mazzola M. Kinetic study of the cure reaction of unsaturated polyester resins. J Therm Anal. 1985;30:1359–66.
Shin SH, Suh MH, Lee SH, Yie JE, Lee JW. A study on curing kinetics of unsaturated polyester resin. Korean Chem Eng Res. 1987;25(5):522–7.
Ng H, Manas-Zloczower I. A nonisothermal differential scanning calorimetry study of the curing kinetics of an unsaturated polyester system. Polym Eng Sci. 1989;29(16):1097–102.
Ramis X, Salla JM. Effect of the inhibitor on the curing of an unsaturated polyester resin. Polym. 1995;36(18):3511–21.
Salla JM, Ramis X. Comparative study of the cure kinetics of an unsaturated polyester resin using different procedures. Polym Eng Sci. 1996;36(6):835–51.
Zetterlund PB, Johnson AF. A new method for the determination of the Arrhenius constants for the cure process of unsaturated polyester resins based on a mechanistic model. Thermochim Acta. 1996;289:209–21.
Yun YM, Lee SJ, Lee KJ, Lee YK, Nam JD. Composite cure kinetic analysis of unsaturated polyester free radical polymerisation. J Polym Sci, Part B: Polym Phys. 1997;35:2447–56.
Martin JL, Cadenato A, Salla JM. Comparative studies on the non-isothermal DSC curing kinetics of an unsaturated polyester resin using free radicals and empirical models. Thermochim Acta. 1997;306:115–26.
Yousefi A, Lafleur PG, Gauvin R. The effects of cobalt promoter and glass fibres on the curing behaviour of unsaturated polyester resin. J Vinyl Addit Technol. 1997;3(2):157–69.
Yousefi A, Lafleur PG, Gauvin R. Kinetic studies of thermoset cure reactions: a review. Polym Compos. 1997;18(2):157–68.
Lu MG, Shim MJ, Kim SW. Curing behavior of an unsaturated polyester system analyzed by Avrami equation. Thermochim Acta. 1998;323:37–42.
Ramis X, Salla JM. Effect of the initiator content and temperature on the curing of an unsaturated polyester resin. J Polym Sci, Part B: Polym Phys. 1999;37:751–68.
Salla JM, Cadenato A, Ramis X, Morancho JM. Thermoset cure kinetics by isoconversional methods. J Therm Anal Calorim. 1999;56:771–81.
Vilas JL, Laza JM, Garay MT, Rodriguez M, Leon LM. Unsaturated polyester resins cure: Kinetic, rheologic, and mechanical-dynamical analysis. I. Cure Kinetics by DSC and TSR. J Appl Polym Sci. 2001;79:447–457.
Cook WD, Lau M, Mehrabi M, Dean K, Zipper M. Control of gel time and exotherm behaviour during cure of unsaturated polyester resins. Polym Int. 2001;50:129–34.
Rouison D, Sain M, Couturier M. Resin transfer moulding of natural fibre reinforced plastic. I. Kinetic study of an unsaturated polyester resin containing an inhibitor and various promoters. J Appl Polym Sci. 2003;89:2553–2561.
Rouison D, Sain M, Couturier M. Resin transfer molding of natural fibre reinforced composites: cure simulation. Compos Sci Technol. 2004;64:629–44.
Boyard N, Sinturel C, Vayer M, Erre R. Morphology and cure kinetics of unsaturated polyester resin/block copolymer blends. J Appl Polym Sci. 2006;102:149–65.
Worzakowska M. Kinetics of the curing reaction of unsaturated polyester resins catalyzed with new initiators and a promoter. J Appl Polym Sci. 2006;102:1870–6.
Martin JL. Kinetic analysis of two DSC peaks in the curing of an unsaturated polyester resin catalysed with methylethylketone peroxide and cobalt octoate. Polym Eng Sci. 2007;47(1):62–70.
Gao J, Dong C, Du Y. Nonisothermal curing kinetics and physical properties of unsaturated polyester modified with EA-POSS. Int J Polym Mater. 2010;59:1–14.
Janković B. The kinetic analysis of isothermal curing reaction of an unsaturated polyester resin: Estimation of the density distribution function of the apparent activation energy. Chem Eng J. 2010;162:331–40.
Kuppusamy RRP, Neogi S. Influence of curing agents on gelation and exotherm behaviour of an unsaturated polyester resin. Bull Mater Sci. 2013;36(7):1217–24.
Poorabdollah M, Beheshty MH, Atai M, Vafayan M. Cure kinetic study of organoclay-unsaturated polyester resin nanocomposites by using advanced isoconversional approach. Polym Compos. 2013;34(11):1824–31.
Aktas A, Krishnan L, Kandola B, Boyd SW, Shenoi RA. A cure modelling study of an unsaturated polyester resin system for the simulation of curing of fibre-reinforced composites during the vacuum infusion process. J Compos Mater. 2014;49(20):2529–40.
S Subaşı S, Poyraz B, Eren Ş, Gökçe N, Aykanat B. Initiator effect on chemical, thermal and mechanical Properties of polyester based composites. In: Proceedings of the 2nd international conference on civil and environmental engineering, Cappadocia, Nevşehir, 2017.
Vyazovkin S, Sbirrazzuoli N. Isoconversional kinetic analysis of thermally stimulated processes in polymers. Macromol Rapid Commun. 2006;27:1515–32.
Vyazovkin S, Burnham AK, Criado JM, Perez-Maqueda LA, Popescu C, Sbirrazzuoli N. Review: ICTAC kinetics committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Vyazovkin S. Isoconversional kinetics of thermally stimulated processes. Springer, New York; 2015.
Sbirrazzuoli N. Determination of pre-exponential factors and of the mathematical functions f(α) or G(α) that describe the reaction mechanism in a model-free way. Thermochim Acta. 2013;564:59–69.
Roduit B, Folly P, Berger B, Mathieu J, Sarbach A, Andres H, Ramin M, Vogelsanger B. Evaluating sadt by advanced kinetics-based simulation approach. J Therm Anal Calorim. 2008;93(1):153–61.
Winter HH. Can the gel point of as cross-linking polymer be detected by the G′–G′′ crossover? Polym Eng Sci. 1987;27(22):1698–702.
Sbirrazzuoli N, Mititelu-Mija A, Vincent L, Alzina C. Isoconversional kinetic analysis of stoichometric and off-stoichometric epoxy-amine cures. Thermochim Acta. 2006;447:167–77.
Alonso MV, Oliet M, García J, Rodríguez F, Echeverría J. Gelation and isoconversional kinetic analysis of lignin-phenol-formaldehyde resol resins cure. Chem Eng J. 2006;122:159–66.
Galwey AK, Brown ME. Kinetic background to thermal analysis and calorimetry. In: Brown ME (Ed.). Handbook of thermal analysis and calorimetry, volume 1: principles and practice. Amsterdam: Elsevier; 1998.
Vyazovkin S. Modification of the integral isoconversional method to account for variation in the activation energy. J Comput Chem. 2001;22(2):178–83.
Friedman HL. Kinetics of thermal degradation of char-forming plastics from thermogravimetry: application to a phenolic plastic. J Polym Sci. 1964;C6:183–195.
Flynn JH. A general differential technique for the determination of parameters for d(α)/dt = f(α) A exp(-E/RT). J Therm Anal. 1991;37(2):293–305.
Galwey AK, Brown ME. Application of the Arrhenius equation to solid state kinetic: can this be justified? Thermochim Acta. 2002;386:91–8.
Vyazovkin S. Model-free kinetics. Staying free of multiplying entities without necessity. J Therm Anal Calorim. 2006;83(1):45-51.
Vyazovkin S, Wight CA. Model-free and model-fitting approaches to kinetic analysis of isothermal and nonisothermal data. Thermochim Acta. 1999;340–341:53–68.
Kim SY, Choir DG, Yang SM. Rheological Analysis of the gelation behavior of tetraethylorthosilane/vinyltriethoxysilane hybrid solutions. Korean J Chem Eng. 2002;19(1):190–6.
ASTM E1545-11 (2016). Standard test method for assignment of the glass transition temperature by thermomechanical analysis.
Kamal MR. Thermoset characterization for moldability analysis. Polym Eng Sci. 1974;14:231–9.
Sourour S, Kamal MR. Differential scanning calorimetry of epoxy cure: Isothermal cure kinetics. Thermochim Acta. 1976;14:41–59.
Vyazovkin S, Wight CA. Isothermal and non-isothermal kinetics of thermally stimulated reactions of solids. Int Rev Phys Chem. 1998;17(3):407–33.
Sbirrazzuoli N, Vyazovkin S, Mititelu A, Sladic C, Vincent L. A study of epoxy-amine cure kinetics by combining isoconversional analysis with temperature modulated DSC and dynamic rheometry. Macromol Chem Phys. 2003;204(15):1815–21.
Acknowledgements
This scientific work was funded by the Austrian Ministry for Transport, Innovation and Technology in frame of the program “Produktion der Zukunft” under Contract No. 858688.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wuzella, G., Mahendran, A.R., Beuc, C. et al. Isoconversional cure kinetics of a novel thermosetting resin based on linseed oil. J Therm Anal Calorim 142, 1055–1071 (2020). https://doi.org/10.1007/s10973-020-09529-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-09529-7