
Abstract
The pharmaceutical cocrystal of salicylic acid (C7H6O3 or H2Sal) and salicylamide (C7H7NO2 or SAM) was synthesized and characterized by various techniques. The differential scanning calorimetry results confirmed the eutectic fusion showing the characteristic of the endothermic sharp peak of the solidus temperature at 108 °C. X-ray crystal structure of cocrystal is orthorhombic with space group Pna2(1). Cocrystal consists of H2Sal and a distorted phenolic group of SAM. The packing diagram of cocrystal H2Sal·SAM clearly confirmed the \(R_{8}^{2}\) acid–amide dimer heterosynthons and other inter- and intramolecular interaction bonds to stabilize the structure. In addition, the strength of the hydrogen bonds is studied using the vibrational spectral measurements, confirming the band shifting due to the intermolecular interactions. The identity of compounds by matching the absorbance spectrum was confirmed by ultraviolet spectroscopy technique. Furthermore, the experimental studies were supported by calculation results using density functional B3LYP methods with the standard 6-311++G(d,p) basis set level. The parameters such as bond lengths, bond angles and Mulliken atomic charges values have been calculated and compared, confirmed the interactions and charge transfers. The frontier molecular orbitals (HOMO–LUMO) illustrated the lower band-gap value suggesting the possible pharmaceutical activity of this as obtained H2Sal·SAM cocrystal.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pharmaceutical cocrystallization is often considered for the development of API drugs [1,2,3,4,5,6,7,8,9,10,11]. This method is about searching the suitable solid form which can enhance the specific physical properties such as solubility, bioavailability, stability, etc. without affecting the biological activity of the drug [1,2,3,4,5,6,7,8,9]. In general, cocrystals are formed through noncovalent interaction bonds (primarily hydrogen bonding), π–π stacking, van der Waals interactions and other weak interactions [1,2,3,4,5,6,7,8,9,10,11].
Salicylic acid or 2-hydroxybenzoic acid (C7H6O3 or H2Sal) is nonsteroid anti-inflammatory drug (NSAID) that shows analgesic effects [12,13,14,15]. Salicylamide 2-hydroxybenzoic acid (C7H7NO2 or SAM) is not only the salicylic derivative but also the NSAID. It is the weak antifungal, antibacterial actions and keratolytic [2, 9, 15]. Salicylic acid and salicylamide are phenolic compounds in which both contain the phenolic hydroxyl group (Ph-OH) [2]. Moreover, carboxyl (C(O)–OH) group of salicylic acid and carboxamide (C(O)–NH2) group of salicylamide are the key tools to generate cocrystal via the concept of supermolecular sythons such as carboxylic–carboxylic, amide–salicylamide and carboxylic–salicylamide denoted as types (I), (II) and (III), respectively (see Fig. 1) [3,4,5,6,7,8,9,10,11].

The chemical structures of H2Sal and SAM and some probable molecular synthons
The cocrystal of salicylic acid and salicylamide has been reported with other molecules for the purpose of increasing pharmaceutical physicochemical properties [3, 9, 10, 15, 16]. The example cocrystals of salicylic acid are with ethenzamide [3] and benzamide [9]. The cocrystals of salicylamide are with itself as homosynthon [10, 15], 3,5-dinitrobenzoic acid [4] and piperidine-3-carboxylic acid [16].
In addition, the pharmaceutical individual crystals and cocrystals of salicylic acid and salicylamide have been studied in detail not only experimentally but also from a theoretical point of view [3, 4, 10, 15,16,17,18,19,20,21]. Computational chemistry is especially used as a tool to understand the physiochemical properties, such as the weak interaction, molecular synthons or motifs as shown in Fig. 1 [10, 21, 22].
However, to the best of our knowledge, there is no report on both preparation and computational chemistry calculations of such cocrystal of salicylic acid and salicylamide (H2Sal·SAM). Therefore, the main objective of this work is to combine the experimental and theoretical investigations for understanding the preparation, molecular interactions and other relative properties and aiming for a drug design.
Experimental
Preparation of compounds
All the chemicals were obtained commercially and used without further purification. For better understanding and accurate data, we recrystallized the starting materials H2Sal and SAM.
H2Sal was obtained by 5 mL of methanol solvent-drop grinding of salicylic acid (0.14 g, 0.01 mol). When the solution was allowed to evaporate to dryness, crystals were obtained within a day with 85% yield.
SAM was also obtained by 5 mL of methanol solvent-drop grinding of salicylamide (0.27 g, 0.02 mol). When the solution was allowed to evaporate to dryness, crystals emerged after 2 days with 80% yield.
Cocrystal H2Sal·SAM: Actually, several attempts were also done to obtain cocrystals. We have tried multiple methods to purify or prepare pharmaceutical compounds such as slow solvent evaporation, cooling, solvent-drop grinding [23] and also slurring techniques [24]. Fortunately, the methods reported in this work gave the single crystals which were suitable for X-ray diffraction studies.
In the typical procedure, pharmaceutical cocrystal H2Sal·SAM was synthesized in the 1:2 mole ratio by the solvent evaporation method. Salicylic acid (0.14 g, 0.01 mol) and salicylamide acid (0.27 g, 0.02 mol) were dissolved in 20 mL of ethanol and continuously stirred for 1 h at room temperature. Then, the solution was filtered off and allowed to slowly evaporate at ambient temperature; single cocrystals were obtained within a week with 83% yield.
Physical measurements
The elemental analyses for C, H, and N were performed on a Perkin-Elmer 240 analyzer. The FT-IR spectra were recorded as KBr pellets on a Perkin-Elmer FT-IR spectrometer in the 4000–400 cm−1 region.
Differential scanning calorimetry (DSC) analysis was carried out using a DSC 204 F1 Phoenix differential scanning heat flux calorimeter (NETZSCH, Germany) with a high sensitivity m-sensor. Sample mass was usually in the range of 5–9 mg. The heating rate is about 10 °C min−1 in argon gas and cooled with liquid nitrogen gas. The UV–Visible spectra are recorded using a Shimadzu UV-1700 spectrophotometer which covered the range of 500–200 nm.
Single-crystal X-ray structure determination
X-ray diffraction studies were carried out using a Bruker APEX2 CCD diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The SAINT program of the SHELXTL software package was used for data collection, cell refinement and data reduction [25]. The SADABS program applied only cocrystal refinement. The structures were solved by direct methods using Shelxs97, and the structure refined against F2 for all reflections by full-matrix methods. The phenolic oxygen atom was examined for the dynamic disorder and then restrain. However, all non-hydrogen atoms were refined anisotropically and the hydrogen atoms were placed in geometric positions and refined as the riding model atoms with C–H = 0.93 Å and Uiso(H) = 1.2 Ueq(C). Program Mercury and Diamond were used to demonstrate the figures [26].
Computational details
For the accuracy, precision and reliability, the initial structure geometries were started from experimental (single-crystal X-ray structure) data. In order to obtain the stable conformer, the self-consistent field (SCF) energy calculation is performed with the HF and DFT methods. From SCF energy calculation, we assumed that the most stable conformer structure is the same as the obtained molecules from X-ray structure determination. We found that DFT calculations with the hybrid density functional method 6-311++G(d,p) are the suitable method (see Table 2), for the prediction and evaluation of organic molecules, which matched well with many literature reviews [9, 10, 15,16,17,18,19,20,21,22].
This hybrid density functional method from the Gaussian 98 program is expected to provide sufficient quantitative accuracy for present purposes [27]. The optimized structure parameters were also used in the computed vibration frequency at the same level to characterize all stationary points as minima. The natural bond orbital (NBO) calculation was also carried out at the same level to get more detailed information about the chemical bonds of the molecule. The frontier molecular orbitals (FMO) has been computed and analyzed. The molecular orbitals were generated using the GaussView program in Gaussian 98 software package [27].
Results and discussion
Experimental pharmaceutical cocrystal H2Sal·SAM
H2Sal, SAM and cocrystal H2Sal·SAM were obtained. For the cocrystal H2Sal·SAM, we successfully prepared the crystals from the 1:2 (salicylic acid: salicylamide) mole ratio with solution crystallization after we have been tried several reactions by changing the preparation methods such as solid–solid grinding [6, 7, 23], solvent-drop grinding with varying the mole ratios [2, 3, 6, 7]. This is to confirm that the solvent evaporation method can be broken and reformed under this condition.
The crystals of H2Sal, SAM and H2Sal·SAM exhibit the colorless needle-like, colorless plate-like and long plate-like morphologies, respectively, as shown in Fig. 2.

The crystal morphology of H2Sal (left), SAM (middle) and H2Sal·SAM (right)
Experimental DSC results
DSC is used as the tool to understand the cocrystalline phase characterization and identification. The DSC thermogram of cocrystal and its starting materials H2Sal and SAM are illustrated in Fig. 3. The DSC curve of cocrystal exhibits the endothermic peaks, similar to those in cocrystal of salicylamide with acetamidobenzoic acid and other similar cocrystals [3, 5,6,7,8,9,10,11].

The DSC curve of cocrystal H2Sal·SAM
The intense and sharp endothermic peak confirm the cocrystal changing from solid to liquid state at 108 °C (enthalpy = 162.00 J g−1), as the melting point of the solid. The cocrystal melting which is lower temperature than those of their starting materials (155 °C of H2Sal and 137 °C of SAM), confirming the eutectic fusion [6, 7, 9, 28, 29]. Moreover, the crystallinity of this process is about 100%, supporting the solubility [9, 30].
Experimental Single-crystal structure of pharmaceutical cocrystal H2Sal, SAM and H2Sal·SAM
The molecular structures of H2Sal, SAM and H2Sal·SAM are shown in Fig. 4. The crystal structure parameters are listed in Table 1.

The structure of H2Sal (left), SAM (middle) and cocrystal H2Sal·SAM (right) plot at approximately 30% with atom-numbering scheme
H2Sal has been previously reported in both experimental and theoretical studies [13, 18]. In this work, H2Sal crystallizes in monoclinic crystal system with a space group P21/c and appears as ortho hydroxyl benzoic acid which is in a different form from the previous reports but in the same form with some experimental reports and the most stable optimization structures.
SAM is also crystallized in the same crystal system and a space group as H2Sal [14]. The replacement of amide (NH2) group to O–H of COOH group yields the SAM structure.
H2Sal·SAM is a cocrystal which crystallizes in orthorhombic crystal system with a space group Pna21. As seen, the phenolic hydroxyl (Ph-OH) is disordered which may effect in hydrogen bond interactions and the rest of the molecules. This phenomenon clearly confirmed by DSC results.
Moreover, the apparent of Flack’s absolute structure parameter (0.8(18)) confirms that cocrystal has chirality which is good for the pharmaceutical selectivity and activity [9].
Experimental and DFT calculation results of structure parameters
Bond, angles and torsion angles of these three crystals structures
Selected bond lengths, bond angles and torsion angles from experimental and theoretical studies are listed in Table 2. The hydrogen bond and weak interactions are shown in Table 3 and Figs. 5–7.

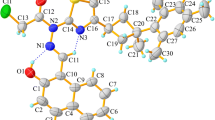
The \(R_{8}^{2}\) acid–acid dimer homosynthon of salicylic acid showing the numbering of the asymmetric unit in the crystal
Indeed, H2Sal consists of three organic functional groups: a carboxylic (COOH), phenolic hydroxyl (Ph-OH) groups and an aromatic cycle. In this work, the carboxylic C=O bond distances are about 0.05(2) Å shorter than the phenolic hydroxyl distances (1.3066(16) Å vs. 1.3066(16) Å). In contrast, the hydroxyl group has longer bond distances (0.96(2)Å vs. 0.93(2) Å).
For SAM, the phenolic hydroxyl (Ph-OH), the carboxylic C=O bond distances are about the same as those in H2Sal.
As seen in Table 2, the structure parameters such as bond lengths, bond angles and torsion angles of cocrystal are shorter or lower than its starting materials, confirming the cocrystal stability than free molecules, for example, the 1.3066(16) Å vs. 1.302(4) Å carboxylic C=O bonds [9].
The hydrogen synthons and weak interactions
As seen in Table 3, the experimental weak interactions of these three compounds are summarized, except the last column of calculated d(H···A), and the experimental structure is illustrated in Figs. 5–7. Undoubtedly, hydrogen bonding plays the important roles to form the building block units known as supramolecular synthons. The calculated weak interactions in all structures are systematic higher than experimental results, because of the gas-phase calculations.
The packing diagram revealed that H2Sal has not only intramolecular hydrogen bonding (O13–H13···O12 of 1.779 Å) but also intermolecular hydrogen bonding (O11–H11···O12 of 1.698 Å) which is similar to the previous reports [3, 6,7,8,9,10]. The intermolecular interaction was \(R_{8}^{2}\) acid–acid dimer homosynthon which is a typical form for salicylic acid [3], as shown in Fig. 5.
Surprisingly, the packing diagram of SAM also revealed the \(R_{8}^{2}\) amide–amide dimer homosynthon and the \(R_{12}^{4}\) amide–amide tetramer homosynthon which has not stated in the previous report [3, 10, 11]. The \(R_{12}^{4}\) tetramer synthon formed through N11–H11···O12 of 2.455 Å intermolecular hydrogen bonds. Overall intra- and intermolecular interaction hydrogen bonding is longer than those of H2Sal as listed in Table 3 and Fig. 6.

The \(R_{8}^{2}\) amide–amide dimer and \(R_{12}^{4}\) amide–amide tetramer homosynthons of salicylamide showing the numbering of the asymmetric unit in the crystal
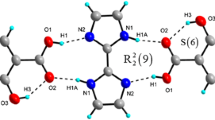
The supramolecular heterosynthon occurred in cocrystal H2Sal·SAM which is the \(R_{8}^{2}\) acid–amide dimer heterosynthons. In this case, the dimer motif is augmented by the amide–acid dimer established through N11–H11···O22 of 1.820 Å and O21–H21···O11 of 2.105 Å and O21–H21···O11 of 2.105 Å. The phenolic O–H···O intramolecular hydrogen bonding of H2Sal is 0.24 Å shorter than that of SAM, as shown in Fig. 7 and Table 3.

The \(R_{8}^{2}\) acid–amide dimer heterosynthon of cocrystal showing the numbering of the asymmetric unit in the crystal
Experimental and DFT calculation results of FT-IR spectra
In order to understand weak interactions, FT-IR spectroscopy is the perfect tool. For the extension of our work, H2Sal, SAM and H2Sal·SAM have been performed by DFT with 6-311++G(p,d). As the results shown in Figs. 8 and 9, the theoretically computed values are typically higher than those of experimental data because of gas-phase calculation which is in very good agreement with experimental results and the previous studies of salicylic acid have been reported [17,18,19,20].

FT-IR spectra of experimental H2Sal, SAM and cocrystal (H2Sal·SAM)

FT-IR spectra of DFT calculation of H2Sal, SAM and cocrystal (H2Sal·SAM)
Actually, the crystal structure of H2Sal contains only three major functional groups which are carboxylic groups, aromatic hydrogen and phenolic functional groups, whereas SAM consists of the primary amide group instead of hydroxyl group [8, 17]. H2Sal·SAM has the combination of H2Sal and SAM functional groups.
Carboxylic group
Carboxylic group (–COOH) consists of carbonyl (C=O) and hydroxyl (O–H) groups. For the O–H vibration, it is the key role and extremely important to form the hydrogen bonding, indicating the broad band between 3600 and 3300 cm−1.
In this work, all the experimental spectra have the very broad peak of carboxylic acid covering the region of 2500–3500 cm−1 indicating the dimer hydrogen bonds [3, 8, 9, 15, 17], supporting the single-crystal X-ray structures data. The overriding spectra are normally centered at around 3000 cm−1. This is to confirm that H2Sal, SAM can form not only monomer but also dimer molecules as the \(R_{8}^{2}\) acid–acid dimer of H2Sal covering the region of 2500–3500 cm−1 indicating the dimer hydrogen bonds [3, 9, 15, 17], supporting the single-crystal X-ray structures data. The overriding spectral is normally centered at around 3000 cm−1. This is to confirm that H2Sal, SAM can form not only monomer but also dimer molecules as the \(R_{8}^{2}\) amide–amide dimer homosynthons SAM [3, 17, 19, 21].
Surprisingly, the experimental spectra of cocrystal H2Sal·SAM appear at the weak broad band in the region of 2300–2800 which is lower than those of free H2Sal and SAM, strongly confirm the \(R_{8}^{2}\) acid–amide dimer heterosynthons of cocrystal H2Sal·SAM. In sum, during the cocrystallization, the band was shifted to the lower band.
The O–H in-plane bending and out of the plane bending vibrations of H2Sal, SAM and H2Sal·SAM appear in the general region of 1380–1440 cm−1 and 549–1179 cm−1. Surprising, the O–H in-plane bending of the dimer wavenumbers are increasing, may be affected by the carboxyl group.
For the carbonyl (C=O) group, the stretching vibration of H2Sal, SAM and H2Sal·SAM are 1652 cm−1, 1689 cm−1 and 1678 cm−1 which are in the region 1650–1715 cm−1 [15]. As the normal expectation, the wavenumber of cocrystals is lower than those of free H2Sal and SAM, due to the intermolecular interactions, appearing at the bridging mode of hydrogen bond [15,16,17,18,19,20].
Phenolic functional groups
The experimental phenolic O–H in-plane bending vibrations of H2Sal and SAM are in the typical wavenumbers of 1100 cm−1–1250 cm−1 [15,16,17,18,19,20,21]. In the cocrystal, the signal appears at 1119 cm−1. There are the peaks at lower than 500 cm−1 (462 cm−1 of H2Sal, 453 cm−1 of SAM) which are identified and assigned to the combination of CO···H and ν(O···H), the corresponding theoretical peaks are at 442 cm−1 of H2Sal, 433 and 415 cm−1 of SAM and 453 cm−1 of cocrystals [17, 21, 22].
Aromatic hydrogen group
The observed spectra peaks appearing at 3097 cm−1, 3192 cm−1 and 3192 cm−1 are corresponding to aromatic C–H stretching vibration of H2Sal, SAM and H2Sal·SAM, respectively [9, 19]. The peaks during 675 cm−1–900 cm−1 belong to C–H ‘oop’ which are 756 cm−1 (H2Sal), 753 cm−1 (SAM) and 752 cm−1 (H2Sal·SAM) [9].
Primary amide group
The observed primary amide of SAM and H2Sal·SAM has two peaks which are asymmetric (3773 cm−1 vs. 3411 cm−1) and symmetric vibrations (3621 cm−1 vs. 3356 cm−1) [9, 15, 19, 21]. The significant decrease in asymmetric amide in cocrystal extremely confirmed this amide group is involving with the N–H···O bonding [9], creating \(R_{8}^{2}\) acid–amide heterodimer of (H2Sal·SAM). The N–H bending peaks are typical in the region of 1650–1560 cm−1, with 1624 cm−1 and 1610 cm−1 (SAM) and 1654 cm−1 and 1612 cm−1 (H2Sal·SAM).
Experimental and DFT calculation results of ultraviolet (UV) spectra
UV spectroscopy was performed in order to identify the functional group or confirm the identity of compounds by matching the absorbance spectrum. All UV spectra of H2Sal, SAM and H2Sal·SAM show the intense at lower wavelengths and weak peaks at higher wavelengths which are 233 and 303 (H2Sal), 231 and 303 nm (SAM) and 223 and 306 nm (H2Sal·SAM). These peaks are corresponded with \(n \to \sigma^{ * }\) and \(\pi \to\pi *\) [4, 31]. The comparison of experimental results is shown in Table 4. Surprisingly, these experimental phenomena indicate about the same wavelengths, supporting an unchanged structure. The calculated experimental energies of \(n \to \sigma^{ * }\) and \(\pi \to\pi *\) are illustrated in the range of 121.67–128.21 kcal mol−1 and 93.44–94.36 kcal mol−1, respectively.
DFT calculation results of HOMO–LUMO energy
Molecular energy from the optimization energy at B3LYP/6-311++G(d,p) level may give us the idea of some properties, such as stability. By comparing the energy of H2Sal and SAM, the energy values of H2Sal is more negative than that of SAM. So, it could be realized that H2Sal is more stable than that of SAM which is corresponding to the higher R-factors of SAM as seen in Tables 1 and 5. The total optimized energy of each free molecules of H2Sal and SAM values is only − 1.021 kcal/mol higher than cocrystal values, assuming a minimal energy for weak interaction to stabilize the cocrystal H2Sal·SAM.
The frontier molecular orbitals (FMOs) consists of the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO), and the (HOMO–LUMO) band-gap energy of H2Sal, SAM and H2Sal·SAM in atomic unit (a.u) is listed in Table 6. The molecular orbital energy may provide information on the chemical activity of the molecule, especially the ground and excited-state proton transfer processes [21]. The energy difference between the HOMO and LUMO or HOMO–LUMO gap is generally the lowest energy electronic excitation that is possible in a molecule.
The HOMO and LUMO of H2Sal, SAM and H2Sal·SAM are corresponding to the π orbitals whose phases are quite different which is similar to other works [21, 24]. As known, the LUMO is an electron acceptor while HOMO is electron donor [21]. In this work, the energies of the HOMO and LUMO are − 0.24577 and − 0.07154 a.u of H2Sal, − 0.35228 and − 0.23843 a.u of SAM and − 0.23552 and − 0.07492 a.u of H2Sal·SAM. The difference between HOMO and LUMO energies are all negative values as shown in Tables 6 and 7. This is to confirm that the high stability is the large HOMO–LUMO band gap [21]. The lower value of frontier orbital energy gaps indicated intramolecular charge transfer within molecules which influence the biological activity of the molecule [21]. As seen in Table 7, the HOMO of H2Sal, SAM and H2Sal·SAM are the π orbitals in which electrons from the HOMO are donated [21]. The HOMO orbitals have the more electronegative atom in which the energy level is lower than others [24]. Since, the weak band of H2Sal, SAM and H2Sal·SAM in the observed UV spectra corresponding with \(\pi \to\pi *\) are 231 nm, 235 nm and 223 nm which concerned with the transition from HOMO to LUMO are in good agreement.
DFT calculation results of Mulliken population analysis
As known, the Mulliken population charges can allow estimating the partial charges of molecules. To prove, we calculated using B3LYP level of theory with 6-311++G(d,p) basis set. Atomic charge values of optimized H2Sal, SAM and H2Sal·SAM molecules are listed in Table 8, and the atomic charges molecules are shown in Fig. 10.

Mulliken atomic charge of H2Sal, SAM and H2Sal·SAM
H2Sal molecule shows the negative charges only on O11, O12, O13, C13, C15 and C17, while the molecule of SAM shows the negative charges on O11, O12, C11, C13, C15, C16, C17 and N11. Whereas, the negative atomic charge of cocrystal H2Sal·SAM molecules are at O21, O22, O23, C21, C23, C25, C26 and C27 for the part of H2Sal and O11, O12, C13, C14, C16 and N11 for the part of SAM. These charges of some atoms are changing, for example, the negative charge of C11 and positive charge of C11, causing by the delocalized electrons. Undoubtedly, all the hydrogen atoms are positive electron acceptor charges. All negative charges are electron donors, corresponding with the charge transfer bonding capability [33]. The higher positive charges of H11a, H12, H13, H17 and H21 than the other hydrogen atoms plays a crucial role in the formation of the \(R_{8}^{2}\) acid–acid dimer homosynthon, \(R_{8}^{2}\) amide–amide dimer homosynthon, \(R_{8}^{2}\) acid–amide dimer heterosynthon and \(R_{12}^{4}\) tetramer synthon.
Conclusions
The long plate-like morphology of cocrystal of salicylic acid (C7H6O3) and salicylamide (C7H7NO2) or H2Sal·SAM are obtained from the 1:2 mole ratio of salicylic acid using the solution-based method for the first time. The DSC of this cocrystal appears at the endothermic peaks corresponding to melting point at 108 °C. The molecular structures of H2Sal, SAM are typical, whereas cocrystal H2Sal·SAM is unique. The disordered phenolic hydroxyl (Ph-OH) of SAM in cocrystal is confirmed by single-crystal X-ray structure result, supporting an unstable, which affects the hydrogen bonds and interactions and later the complicated preparation. Moreover, these experimental structure results were calculated using DFT with 6-311++G(p,d) calculation to understand properties. The experimental and DFT calculation results are in good agreement. The stability is involved with the hydrogen synthons and weak interactions such as the \(R_{8}^{2}\) acid–acid dimer homosynthon of H2Sal, the \(R_{8}^{2}\) amide–amide dimer homosynthon, the new report of \(R_{12}^{4}\) amide–amide tetramer homosynthon of SAM and the \(R_{8}^{2}\) acid–amide dimer heterosynthons of cocrystal H2Sal·SAM. The dimer phenomenon clearly confirmed the \(R_{8}^{2}\) acid–amide dimer heterosynthons from the broad band at 2300–2800 cm−1 from both experimental and calculated FT-IR results and all of the positive electron acceptor charges from the Mulliken results. The experimental and DFT calculation UV results confirmed the absorbance spectra of \(n \to \sigma^{ * }\) and \(\pi \to \pi *\). The calculated HOMO–LUMO energy gaps indicated the minimal energy change in cocrystal compared to an individual molecule. In addition, the lower band-gap values suggest the possible pharmaceutical activity which is fitted well with the appearance of Flack’s absolute structure parameter (0.8(18)).
Supplementary data
CCDC data 1545141-1545143 contain the supplementary crystallographic data of salicylic acid, salicylamide and cocrystal. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Brittain HG. Cocrystal systems of pharmaceutical interest: 2010. Cryst Growth Des. 2012;12:1046–54.
Jones W, Samuel Motherwell WD, Trask AV. Pharmaceutical cocrystals: an emerging approach to physical property enhancement. MRS Bull. 2006;31:875–9.
Aitipamula S, Wong ABH, Chow PS, Tan RBH. Pharmaceutical cocrystals of ethenzamide: structural, solubility and dissolution studies. Cryst Eng Commun. 2012;14:8515–24.
Singh M, Rai RN, Rai US. Synthesis, crystal growth and physicochemical studies on a novel organic inter-molecular compound; 3,5-dinitrobenzoic acid and salicylamide system. J Cryst Growth. 2015;419:114–22.
Zhou Z, Chan HK, Sung HH-Y, Tong HHY, Zheng Y. Identification of new cocrystal systems with stoichiometric diversity of salicylic acid using thermal methods. Pharm Res. 2016;33:1030–9.
Manin AN, Voronin AP, Drozd KV, Manin NG, Bauer-Brandl A, Perlovich GL. Cocrystal screening of hydroxybenzamides with benzoic acid derivatives: a comparative study of thermal and solution-based methods. Eur J Med Sci. 2014;65:56–64.
Surov AO, Manin AN, Voronin AP, Churakov AV, Perlovich GL, Vener MV. Weak interactions cause packing polymorphism in pharmaceutical two-component crystals. The case study of the salicylamide cocrystal. Cryst Growth Des. 2017;17:1425–37.
Évora AOL, Castro RAE, Maria TMR, Ramos Silva M, ter Horst JH, Canotilho J, Eusébio MES. A thermodynamic based approach on the investigation of a diflunisal pharmaceutical co-crystal with improved intrinsic dissolution rate. Int J Pharm. 2014;466:68–75.
Elbagerma MA, Edwards HGM, Munshi T, Scowen IJ. Identification of a new co-crystal of salicylic acid and benzamide of pharmaceutical relevance. Anal Bioanal Chem. 2010;397:137–46.
Manin AN, Voronin AP, Manin NG, Vener MV, Shishkina AV, Lermontov AS, Perlovich GL. Salicylamide cocrystals: screening, crystal structure, sublimation thermodynamics, dissolution, and solid-state DFT calculations. J Phys Chem B. 2014;118:6803–14.
Seato CC, Parkin A. Making benzamide cocrystals with benzoic acids: the influence of chemical structure. Cryst Growth Des. 2011;11:1502–11.
Nordstrom FL, Rasmuson AC. Solubility and melting properties of salicylic acid. J Chem Eng Data. 2006;51:1668–71.
Nordstrom FL, Rasmuson AC. Solubility and melting properties of salicylamide. J Chem Eng Data. 2006;51:1775–7.
Sasada Y, Takano T, Kakudo M. Crystal structure of salicylamide. Bull Chem Soc Jpn. 1964;37(7):940–6.
Arjunan V, Kalaivani M, Ravindran P, Mohan S. Structural, vibrational and quantum chemical investigations on 5-chloro-2-hydroxybenzamide and 5-chloro-2-hydroxybenzoic acid. Spectrochim Acta Part A. 2011;79:1886–95.
Bartoszek-Adamska E, Dega-Szafran Z, Krociak M, Jaskolski M, Szafran M. Hydrogen bonds in 1:1 complex of piperidine-3-carboxylic acid with salicylic acid. J Mol Struct. 2009;920:68–74.
Boczar M, Boda L, Wojcik MJ. Theoretical modeling of infrared spectra of hydrogen-bonded crystals of salicylic acid. Spectrochim Acta A. 2006;64:757–60.
Munshi P, Guru Row TN. Intra- and intermolecular interactions in small bioactive molecules: cooperative features from experimental and theoretical charge-density analysis. Acta Cryst. 2006;B62:612–26.
Esrafili MD. Intra- and inter-molecular interactions in salicylic acid -theoretical calculations of 17O and 1H chemical shielding tensors and QTAIM analysis. Can J Chem. 2011;89:1410–8.
Kwon Y. Theoretical study on salicylic acid and its analogues: intramolecular hydrogen bonding. J Mol Struct (Theochem). 2000;532:227–37.
Karthick T, Balachandran V, Perumal S, Nataraj A. Spectroscopic studies, HOMO–LUMO and NBO calculations on monomer and dimer conformer of 5-nitrosalicylic acid. J Mol Struct. 2011;1005:192–201.
Velcheva EA, Stamboliyska BA. Structural changes caused by the conversion of 2-hydroxybenzamide salicylamide into the oxyanion. J Mol Struct. 2008;875:264–71.
Trask AV, Jones W. Crystal engineering of organic cocrystals by the solid-state grinding approach. Top Curr Chem. 2005;254:41–70.
Karabacak M, Sinha L, Prasad O, Cinar Z, Cinar M. The spectroscopic (FT-Raman, FT-IR, UV and NMR), molecular electrostatic, potential, polarizability and hyperpolarizability, NBO and HOMO–LUMO analysis of monomeric and dimeric structures of 4-chloro-3,5-dinitrobenzoic acid. Spectrochim Acta Part A. 2012;93:33–46.
Sheldrick GM. A short history of SHELX. Acta Crystallogr Sect A. 2008;64:112.
Brandenburg K. Diamond. Bonn: Cryst Impact GbR; 1999.
Frisch J, Trucks GW, et al. Gaussian 98. Pittsburgh(PA): Gaussian Inc; 1998.
Rai US, Singh M, Rai RN. Some physicochemical studies on organic eutectics and intermolecular compounds. J Therm Anal Calorim. 2017;130:967–74.
Fernandes RP, do Nascimento ALCS, Carvalho ACS, Teixeira JA, Ionashiro M, Caires JC. Mechanochemical synthesis, characterization, and thermal behavior of meloxicam cocrystals with salicylic acid, fumaric acid, and malic acid. J Therm Anal Calorim. 2019. https://doi.org/10.1007/s10973-019-08118-7.
Landolt HH. Schmelzgleichgewichte, Vol. II, Part 3. 6th ed. Berlin: Springer; 1950. p. 1831–910.
Babhair SA, Al-Badr AA, Aboul-Enein HY. Salicylamide in analytical profiles of drug substances. In: Florey K, editor. Analytical profiles of drug substances, vol. 13. Orlando: Academic Press; 1984. p. 521–51.
Trivedi MK, Branton A, Trivedi D, Shettigar H, Bairwa K, Jana S. Fourier transform infrared and ultraviolet-visible spectroscopic characterization of biofield treated salicylic acid and sparfloxacin. Nat Prod Chem Res. 2015;3:1–6.
Amalanathan M, Rastogi VK, Hubert Joe I, Palafox MA, Tomar R. Density functional theory calculations and vibrational spectral analysis of 3,5-dinitrobenzoic acid. Spectrochim Acta Part A. 2011;78:1437–44.
Acknowledgements
We gratefully acknowledge the Thaksin University for the research grants via the Research and Development Institute, Thaksin University (RDITSU), and the National Research Management System (NRMS); project codes: 111006 and 184788.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Phetmung, H., Musikapong, K. & Srichana, T. Thermal analysis, structure, spectroscopy and DFT calculations of a pharmaceutical cocrystal of salicylic acid and salicylamide. J Therm Anal Calorim 138, 1207–1220 (2019). https://doi.org/10.1007/s10973-019-08794-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08794-5