
Abstract
Raman spectroscopy, X-ray powder diffraction/X-ray crystallography and differential scanning calorimetry have been used to study the phenomenon of co-crystal formation in stoichiometric mixtures of salicylic acid with benzamide. Raman spectroscopy was particularly useful for the characterization of the products and was used to determine the nature of the interactions in the co-crystals. It was observed that little change in the vibrational modes associated with the phenyl groups of the respective reactants took place upon co-crystal formation, but changes in intensities of the vibrational modes associated with the amide and the carboxylic acid groups were observed upon co-crystal formation. Several new vibrational bands were identified in the co-crystal which were not manifested in the physical mixture of both components and could be used as diagnostic features of co-crystal formation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Co-crystallisation is a crystallisation method that allows the binding of two or more building blocks within one periodic crystalline lattice by hydrogen bonding, π-stacking and van der Waal’s forces [1–3]. The synthesis of crystalline supramolecular structures mediated by hydrogen bonds is of considerable importance [4–7]; the main difference between co-crystals and salts, for example, is that in salts, a proton is transferred from the acidic to the basic functionality of the constituent free base molecule, or vice versa if applicable, whereas in co-crystals, no such transfer occurs [8]. On the other hand, the primary difference between co-crystals and solvates/hydrates is the physical state of the isolated pure components: If one component is a liquid at room temperature, the crystals are designated as solvates/hydrates; if both components are solids at room temperature, the crystals are designated as co-crystals [9].
In the pharmaceutical context, the attraction of investigation of co-crystal forms of drugs relates to four principal factors [10]: design—many active pharmaceutical ingredients (API) contain functional groups predisposed to hydrogen bonding that can be exploited in a cogent way to form pharmaceutical co-crystals; discovery—several processes, including conventional solvent crystallisation, solvent drop grinding and melt crystallisation, can be used to produce diverse ranges of co-crystal forms of API; diversity—pharmaceutical co-crystals exhibit different properties compared with pure crystal forms of API; development—co-crystals represent non-obvious forms of APIs and therefore can represent intellectual property for future exploitation. Recently, the increased interest is on means of optimising the physicochemical properties of solid dosage forms. In addition to potential improvements in solubility, dissolution rate, bioavailability and physical stability, pharmaceutical co-crystals frequently enhance other essential properties of the APIs such as hygroscopicity, chemical stability, compressibility and flowability [11] (Scheme 1).

Molecular structures of salicylic acid (a) and benzamide (b)
Co-crystals can be prepared through evaporation [12] or cooling [13] of a heteromeric solution, co-grinding the components [14], sublimation and growth from the melt [15, 16] or slurry [17, 18]. Solution crystallisation is the preferred method of co-crystal formation, particularly to obtain single crystals for structural analysis [19, 20].
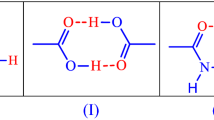
Hydrogen bonding is one of the most important fundamental interactions that cause association of organic molecules, forming the building block units known as supramolecular synthons. One example of a supramolecular homosynthon is the molecular dimer that exists in crystals of carboxylic acids (formation I). However, formation of I is unlikely in competitive situations. Allen et al. [21] determined the probability of formation of 75 bimolecular hydrogen-bonded ring synthons in organic crystal structures. The probability of formation of I was found to be only 33%. This relatively low probability was attributed to competition with other hydrogen-bonded acceptors (e.g. COO−, pyridine N, amide C=O, S=O, P=O).
Another supramolecular synthon of interest would be the molecular dimer that exists in crystals of organic amides which may be typified by benzamide (formation II).
A third supramolecular synthon of original interest is heteromeric hydrogen bond interactions between a phenyl carboxylic acid and a phenyl amide, which is provided by a dimeric species containing O⋅⋅H–N and O–H⋅⋅O hydrogen bonds (formation III).

Ab initio calculations support the idea that the acid–amide supramolecular heterosynthon (III) is favoured over (I) and (II) [22–27]. Also, a survey of hydrogen-bonded co-crystal in the Cambridge Structural Database [28] reveals that most of them have been prepared using strategies that utilise suitable combinations of chemical entities (or functional groups) located on different molecules such that they would prefer to interact and bind heteromerically, formation III, rather than with themselves (homomerically, formations I and II) [29].
These observations are particularly relevant for the design of co-crystals since robust supramolecular heterosynthons represent perhaps the most reliable and rational route to co-crystals. Furthermore, complementary supramolecular heterosynthons that seem to clearly favour formation of co-crystals are not limited to carboxylic acids.
Experimental
Materials
Salicylic acid (SA) and benzamide (BA) were purchased from Sigma-Aldrich at >98% purity. These materials were used as received. The solvent (ethanol) was HPLC grade and purchased from Reidel de Haën or Fisher Scientific.
Co-crystal formation was identified initially using Raman spectroscopy and the difference in melting points between the pure components and the product; the co-crystalline structures were confirmed by X-ray powder diffraction and single-crystal X-ray diffraction.
Co-crystallization via slow evaporation
A 1:1 molar ratio mixture of salicylic acid (100 mg, 0.724 mmol) and benzamide (87.70 mg, 0.724 mmol) was dissolved in 15 mL of ethanol with slight warming until dissolution was practically complete. The filtered solution was then allowed to slowly evaporate at room temperature.
Raman spectroscopy
Raman spectra of the co-crystal samples and those of the single components were obtained using an inVia Raman microscope (Renishaw plc.) with 785-nm stabilised diode laser excitation. The laser power at the sample was approximately 25 mW. A 50× objective lens was used, giving a laser spot diameter (footprint) of about 2 µm at the sample. Spectra were obtained for a 10-s exposure of the CCD detector in the wavenumber region 3,600–50 cm−1 using the extended scanning mode of the instrument.
Powder X-ray diffraction
Data were collected on a Bruker D8 diffractometer in Bragg–Brentano θ–θ geometry with Cu Kα1,2 radiation (1.5418 Å) using a secondary curved graphite monochromator. The X-ray tube was operated at 40 kV, 30 mA. Samples were scanned in a vertical Bragg–Brentano (θ/2θ) geometry (reflection mode) from 5° to 40° (2θ) using a 0.005° step width and a 1.5-s count time at each step. The receiving slit was 1° and the scatter slit 0.2°.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) profiles were generated in the range of −50 to 160 °C using a TA Q2000 DSC instrument with an RGS90 cooling unit. Temperature calibration was performed using an indium metal standard supplied with the instrument at the appropriate heating rate of 10 °C min−1. Accurately weighed samples (1–2 mg) were placed in Tzero aluminium pans using a similar empty pan as reference. The data were collected in triplicate for each sample and were analysed using TA Instruments Universal Analysis 2000 version 4.3A software.
Single-crystal X-ray diffraction
Single-crystal data were collected on a Bruker Apex II CCD diffractometer with Mo Kα radiation (0.71073 Å). The structure was solved by direct methods with SHELXS-97 and refined by a full-matrix least squares analysis on F 2 with anisotropic displacement parameters for non-H atoms in SHELXL-97.
Crystal data for salicylic acid–benzamide co-crystal
C14H13NO4, M = 259.25, monoclinic, a = 5.4523(3) Å, b = 9.6459(7) Å, c = 23.9008(15) Å, α = 90.00°, β = 92.716(2)°, γ = 90.00°, V = 1255.59(14) Å3, T = 296(2) K, space group P21/c, Z = 4, μ(Mo Kα) = 0.102 mm−1, 14,890 reflections measured, 3,583 independent reflections (R int = 0.0211). The final R 1 values were 0.0422 (I > 2σ(I)). The final wR(F 2) values were 0.1156 (I > 2σ(I)). The final R 1 values were 0.0559 (all data). The final wR(F 2) values were 0.1263 (all data) (Table 1).
Result and discussion
The Raman spectra of salicylic acid, benzamide and the mixture are shown in Figs. 1, 2 and 3, and the vibrational wavenumbers and assignments are listed in Table 2. Raman spectroscopic data were utilised first to evaluate whether the complex is a simple physical mixture or a component of molecular ions. Salicylic acid contains a carboxylic group and a hydroxyl group, whereas benzamide contains a carboxyl group and primary amide group. It was found that very little perturbation of the wavenumbers of the carbon–carbon and carbon–hydrogen modes was observed in the fingerprint region of the Raman spectra, indicating that these structures were effectively the same in the co-crystal as they were in the initial reactants. However, the vibrational bands associated with the amide group of benzamide and the bands associated with the carboxyl group of salicylic acid were found to undergo significant changes, as shown in Table 2.

Raman spectra in the 3,120- to 3,000-cm−1 region: a BA, b SA and c SA–BA

Raman spectra in the 1,750- to 1,400-cm−1 region: a BA, b SA and c SA–BA

Raman spectra in the 1,400- to 1,100-cm−1 region: a BA, b SA and c SA–BA
The peak at 3,086 cm−1 (7b (C–H) stretching), 3,075 cm−1 (2 (C–H) stretching and 3,065 cm−1 (C–H) stretching mode in the spectra of salicylic acid and benzamide, respectively, appears as a broad peak centred at 3,070 cm−1 in the spectrum of the mixture, as shown in Fig. 1.
As shown in Figs. 1, 2, 3 and 4 and Table 2, during the formation of a SA–BA crystal, the O–H and C=O bands of salicylic acid and benzamide are shifted to higher or lower wavenumbers by 10 to 30 cm−1, which suggests that the molecular complex of salicylic acid and benzamide is a co-crystal and not simply a mixture of these components [30]. Whilst intramolecular and intermolecular hydrogen bonding modes are shown by both benzamide and salicylic acid [31, 32], the changes in the positions of these bands point to the formation of intermolecular amide–acid interactions as evidenced in the crystal structure of the co-crystal (vide infra).

Raman spectra in the 1,070- to 110-cm−1 region: a BA, b SA and c SA–BA
On the other hand, if the amide carbonyl group of benzamide is involved in an intermolecular hydrogen bond, it would also be expected to shift to a lower wavenumber. As shown in Fig. 2 and Table 2, the Raman spectra for pure benzamide in the starting material have bands at 1,685 and 1,569 cm−1, corresponding to the amide I band and amide II band, respectively. During the co-crystallisation of benzamide with salicylic acid, these bands in the co-crystal were shifted to 1,655 and 1,548 cm−1, respectively. The decrease in amide I frequency of benzamide from 1,685 to 1,655 cm−1 and from 1,569 to 1,548 cm−1 in the SA component indicates that the amide carbonyl group is participating in a strong hydrogen bonding.
The Raman spectra for pure salicylic acid in the starting material have strong bands at 1,632 and 1,322 cm−1 and medium peak at 1,383 cm−1 corresponding to C=O stretching, (C–O)c stretching and (O–H)h in plane bend, respectively. During the co-crystallisation, the band at 1,632 cm−1 was shifted to 1,612 cm−1, the band at 1,383 cm−1 disappears, and the peak at 1,322 cm−1 appears as triplet peaks at 1,331, 1,322 and 1,315 cm−1; these observations indicate that the carboxylic group is participating in strong hydrogen bonding, as shown in Figs. 2 and 3. The peaks in the spectrum of benzamide occurring at 1,179, 1,171 and 1,122 cm−1 can be attributed to an in-plane C–H deformation mode (9a), in-plane C–H deformation mode (7a) and in-plane C–H deformation mode, respectively. In a co-crystal, these bands were shifted to 1,174, 1,164 and 1,115 cm−1, respectively (Figs. 5, 6 and 7).

Raman spectra in the 550- to 200-cm−1 region: a BA, b SA and c SA–BA

Raman spectra in the 1,750- to 1,300-cm−1 region: a BA, b SA, c SA-BA and d SABA 1:1 physical mixture

Raman spectra in the 1,200- to 400-cm−1 region: a BA, b SA, c SA-BA and d SABA 1:1 physical mixture
The (18b(C–C)) in-plane bending band at 1,029 cm−1 in the spectrum of the salicylic acid is shifted to higher wavenumbers and appears at 1,035 cm−1 in the spectrum of the mixture. Moreover, new peaks at 746 and 540 cm−1, which do not occur in either salicylic acid or benzamide, appear in the SA–BA co-crystal. The peaks at 449 cm−1 (9b) mode, 432 cm−1 (16a) mode and 283 cm−1 (C–O)h in-plane bend are shifted to 444, 420 and 276 cm−1, respectively. In the co-crystal, the peaks at 256 cm−1 (10a mode) in the spectrum of the salicylic acid and 242 cm−1 in the spectrum of the benzamide appear as a single peak at 249 cm−1 in the spectrum of the co-crystal. Two new peaks appear in the spectrum of the co-crystal at 189 and 160 cm−1. The Raman spectrum of the SA–BA 1:1 physical mixture is shown in Fig. 8; it can be observed that the physical mixture has very similar band signatures with the raw material (SA and BA). Hence, the Raman data show a clear distinction between the physical mixture and the SA–BA co-crystal.

Powder X-ray diffraction pattern in region (6–20) 2θ: a SA–BA, b SA and c BA
The powder X-ray diffraction (XRD) diffractograms of the ethanol-recrystallised product of the specimen from Figs. 8 and 9 are clearly different from those of the starting materials and the co-crystal product, confirming the presence of a new crystalline phase.

Powder X-ray diffraction pattern in region (20–40) 2θ: a SA-BA, b SA and c BA
The XRD pattern of the co-crystal was compared with the physical mixture (Fig. 10); it can be observed that the co-crystal has a different XRD pattern from the physical mixture. On the other hand, the diffraction pattern for the physical mixture is a superposition of those of the raw materials (Fig. 10).

Powder X-ray diffraction in region (5–30) 2θ: a SA–BA 1:1 physical mixture, b SA–BA co-crystal, c BA and d SA
Furthermore, DSC thermogram of the product exhibits a melting endotherm maximum at 118 °C, providing further support for a new crystal phase (Fig. 11). The thermal behaviour of the new phase is significantly different from salicylic acid (melting endotherm maximum at 160 °C) and benzamide (melting endotherm maximum at 127.8 °C).

DSC melting curves of co-crystal, benzamide and salicylic acid
A single crystal was selected from the product, and X-ray structure determination confirmed the existence of a 1:1 co-crystal formed from salicylic acid and benzamide. The structure of the asymmetric co-crystal (Fig. 12) establishes the stoichiometry of the co-crystal and shows the primary structural motif as an amide–acid dimer established through hydrogen bonding. Bond lengths in the individual molecules of the co-crystal are unremarkable. Amide–acid dimers are well-established motifs in crystal engineering. In this case, the dimer motif is augmented by an intramolecular hydrogen bond in the salicylic acid moiety, formed by the phenolic hydrogen and the carbonyl of the acid group. The crystal packing (Fig. 13) is dominated by chains of dimer units propagating parallel to the b-axis of the cell along the 21-screw axis and formed through hydrogen bonds between the second hydrogen of the amide and the carbonyl of the amide group of the adjacent dimer unit. The characteristics of the hydrogen-bonded network is summarised in Table 3.

Hydrogen-bonded acid–amide dimer motifs in the 1:1 co-crystal of benzamide and salicylic acid showing the numbering of the asymmetric unit in the crystal

Crystal packing in the 1:1 co-crystal of benzamide and salicylic acid viewed in orthographic projection down the a-axis of the unit cell showing chains of dimer units linked through C=O…HN hydrogen bonds
Conclusions
This study establishes that salicylic acid and benzamide form a co-crystal in a 1:1 molar ratio through slow evaporation from ethanol solution. The high selectivity and good interpretational abilities of Raman spectroscopy make it possible to identify spectral contributions from each chemical constituent by a peak wavenumber comparison of single-component spectra (API and guest) and the two-component sample material (API/guest), thus allowing a direct assessment of co-crystal formation to be made. Deductions from interpretation of the Raman spectra have been substantiated by X-ray diffraction and thermal methods.
References
Aakeroy CB, Salmon DJ (2005) Cryst Eng Commun 7:439–448
Morissette SL, Almarsson O, Peterson ML, Remenar JF, Read MJ, Lemmo AV, Ellis S, Cima MJ, Gardner CR (2004) Adv Drug Deliv Rev 56:275–300
Peterson ML, Hickey MB, Zaworotko MJ, Almarsson O (2006) J Pharm Pharmaceut Sci 9:317–326
Jones W (1997) Organic molecular solids: properties and applications. CRC, New York
Atwood JL, Davies JED, MacNicol DD, Vogtle F (1996) Comprehensive supramolecular chemistry, vol 9. Pergamon, Oxford
Bond AD, Jones W (2002) Supramolecular organization and materials design. Cambridge University Press, Cambridge
Desiraju GR (1989) Crystal engineering: the design of organic solids. Elsevier, New York
Banerjee R, Bhatt PM, Ravindra NV, Desiraju GM (2005) Cryst Growth Des 6:2299–2309
Chiarella RA, Davey RJ, Peterson ML (2007) Cryst Growth Des 7:1223–1226
Shan N, Zaworotko MJ (2008) Drug Discov Today 13:440–446
Almarsson O, Zaworotko MJ (2004) Chem Commun 17:1889–1896
Shattock TR, Arora KK, Vishweshwar P, Zaworotko MJ (2008) Cryst Growth Des 8:4533–4545
Hickey MB, Peterson ML, Scoppettuolo LA, Morrisette SL, Vetter A, Guzman H, Remenar JF, Zhang Z, Tawa MD, Haley S, Zaworotko MJ, Almarssono O (2007) Eur J Pharm Biopharm 67:112–119
Chadwick K, Davey R, Cross W (2007) Cryst Eng Commun 9:732–734
Palmer DS, Llinas A, Morao I, Day GM, Goodman JM, Glen RC, Mitchell JBO (2008) Mol Pharm 5:266–279
Seefeldt K, Miller J, Alvarez-Nunez F, Rodrıguez-Hornedo N (2007) J Pharm Sci 96:1147–1158
Zhang GGZ, Henry RF, Borchardt TB, Lou XC (2007) J Pharm Sci 96:990–995
Takata N, Shiraki K, Takano R, Hayashi Y, Terada K (2008) Cryst Growth Des 8:3032–3037
Bis JA, Vishweshwar P, Middleton RA, Zaworotko MJ (2006) Cryst Growth Des 6:1048–1053
Fleishman SG, Kuduva SS, McMahon JA, Moulton B, Rosa D, Bailey W, Rodrıguez-Hornedo N, Zaworotko MJ (2003) Cryst Growth Des 3:909–919
Allen FH, Motherwell WDS, Raithby PR, Shields GP, Taylor R (1999) New J Chem 23:25–34
Arkema S, Bats JW, Weyenberg AM, Feil D (1972) Acta Crystallogr B 28:1646–1648
Leiserowitz L, Nader F (1977) Acta Crystallogr B 33:2719–2733
Reddy LS, Nangia A, Lynch VM (2004) Cryst Growth Des 4:89–94
Aakeroy CB, Beatty AM, Helfrich BA, Nieuwenhuyzen M (2003) Cryst Growth Des 3:159–165
Vishweshwar P, Nangia A, Lynch VM (2002) J Org Chem 67:556–565
Vishweshwar P, Nangia A, Lynch VM (2003) Cryst Growth Des 3:783–790
Allen FA (2002) Acta Crystallogr Sect B 58:380
Aakeroy CB, Beatty AM, Nieuwenhuyzen M, Zou M (2000) Tetrahedron 56:6693–6699
Childs SL, Stahly GP, Park A (2007) Mol Pharm 4:323–338
Blake CCF, Small RWH (1972) Acta Crystallogr B 28:2201–2206
Boczar M, Boda L, Wojcik MJ (2006) Spectrochim Acta Part A 64:757–760
Volovsek V, Colombo L, Furic K (1983) J Raman Spect 14:347–352
Humbret B, Alnot M, Quiles F (1998) Spectrochim Acta Part A 54:465–476
Kniseley RN, Fassel VA, Farquhar EL, Gray LS (1962) Spectrochim Acta 18:1217–1229
Smith BC (1998) Infrared spectral interpretation: a systematic approach. CRC, Boca Raton
Additional Information Available
X-ray crystallographic information for the crystal structures (PDF); this material is available free of charge via the Internet at http://pubs.acs.org. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC-756492. Copies of available material can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: + 44 (0) 1223-336033 or e mail: teched@chemcrys.cam.ac.uk).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 450 kb)
Rights and permissions
About this article
Cite this article
Elbagerma, M.A., Edwards, H.G.M., Munshi, T. et al. Identification of a new co-crystal of salicylic acid and benzamide of pharmaceutical relevance. Anal Bioanal Chem 397, 137–146 (2010). https://doi.org/10.1007/s00216-009-3375-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3375-7