
Abstract
The enzyme, cutinase from Saccharomonospora viridis AHK190 (Cut190), can hydrolyze the inner block of polyethylene terephthalate (PET). Cut190 has a unique feature that both its activity and thermal stability are increased upon Ca2+ binding. In consideration of the glass transition temperature of PET, which is between 60 and 65 °C, the increased activity and thermal stability are of great interest to apply for PET bio-recycling. Our previous mutational analysis showed that the S226P/R228S mutant (Cut190*) has a higher activity and thermal stability than those of the wild type. In this study, we analyzed the folding thermodynamics of the inactive mutant of Cut190*, Cut190*S176A, using circular dichroism and differential scanning calorimetry. The results show that the denaturation temperature increases from 54 to 71 °C due to the addition of 250 mM Ca2+, in a Ca2+ concentration-dependent manner. The increased thermal stability is mainly due to the increased enthalpy change, partially compensated by the increased entropy change. Based on the crystal structure of Cut190*S176A bound to Ca2+, molecular dynamics simulations were carried out to analyze the effects of Ca2+ on the structural dynamics, showing that the Ca2+-bound structure fluctuated less than the Ca2+-free structure. Structural analysis indicates that Ca2+ binding increases the intramolecular interactions of the enzyme, while decreasing its fluctuation, which are in good correlation with the experimental results of the folding thermodynamics.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cutinases (EC 3.1.1.74) belong to a lipase superfamily and were found to catalyze the hydrolysis of complex plant biopolymers in the cuticle layer of leaves (cutin), as well as in the suberin layer of roots (suberin), when cutinases were discovered as a tool of phytopathogenic fungi to invade into the plant surfaces [1, 2]. Because cutinases hydrolyze various substrates such as lipids, waxes, and synthetic esters, as well as catalyzing esterification and transesterification reactions and are produced by fungi, bacteria, and plant pollen [3], they have been used as biocatalysts in industrial applications for the synthesis and bioconversion of chemical compounds [4, 5]. Degradation of polyethylene terephthalate (PET) is of increasing interest as plastic disposal attracts worldwide attention and enzymatic hydrolysis of PET has been evaluated for this purpose [6, 7]. It is noteworthy that surface hydrolysis of PET films or fibers has been observed by serine hydrolases even at temperatures lower than the glass transition temperature (Tg) of PET (which is between 60 and 65 °C in aqueous solution), although significant decay of the polymer structure (especially the inner block leading to significant mass loss) was only detected at temperatures higher than Tg [6, 8]. This is due to the increased mobility of the polymer chain at temperatures higher than Tg, making the approach of an enzyme cleaving the polymer body feasible, although the enzyme has to be thermostable at least at temperatures around Tg. In addition, PET hydrolases have to possess a larger active center that can accommodate an aromatic ring, compared with aliphatic polyester hydrolases. Because of limitations with respect to the substrate specificity and thermostability required for PET hydrolysis, there are still only a few examples of cutinases able to significantly hydrolyze the inner block of PET [8].
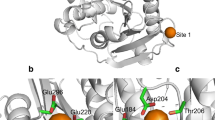
We previously cloned a few cutinases from thermophilic actinomycetes, T. alba AHK119 and Saccharomonospora viridis AHK190 [8,9,10]. Our structural and functional analysis, together with mutational analysis, revealed that a mutant of cutinase from Saccharomonospora viridis AHK190 (Cut190), S226P/R228S, denoted as Cut190*, was the best enzyme for PET degradation given its activity and thermal stability [8]. The enzyme Cut190 has a unique feature that Ca2+ binding increases both the activity and the thermal stability [8], which resulted in an increase in hydrolysis of two PET films by 14 and 27% at 63 °C. Because the PET-hydrolyzing activity increases with increasing temperatures, high thermal stability of Ca2+ bound Cut190* is of interest for both basic science of protein chemistry and industrial applications. Our previous crystal structure analysis of the Cut190 mutant S226P has shown that a Ca2+ ion is coordinated within a loop which induces large conformational changes in three loops corresponding to the β1–β2 loop, the β3–α2 loop, and the β4–α3 loop [11]. Furthermore, recent crystal structure analysis of an inactive mutant, Cut190*S176A, in complex with Ca2+ and a substrate has shown that three Ca2+ ions bind to the protein surface (Fig. 1), one of which is the same site reported previously [11], and regulate the catalysis.

The crystal structure of Cut190*S176A in complex with Ca2+ (PDB ID; 5zno). The blue spheres indicate Ca2+ molecules
In this study, to understand the molecular mechanism of increased stability induced upon Ca2+ binding, we analyzed the folding thermodynamics of Cut190*S176A under various concentrations of Ca2+, using circular dichroism (CD) and differential scanning calorimetry (DSC). We also analyzed the effects of Ca2+ binding on the structural dynamics of Cut190*S176A using molecular dynamics (MD) simulations on both the Ca2+-bound and Ca2+-free structures under various Ca2+ concentrations starting from the corresponding crystal structures. The thermodynamic origin of the increased stability could be explained by the conformational change in Cut190*S176A induced upon Ca2+ binding.
Experimental
Preparation of Cut190*S176A
The expression plasmid of pQE80L-cut190*S176A was used for transformation of E. coli Rosetta-gami B (DE3) cells, as described previously [8]. The protein was expressed under a M9 minimum medium [12], in order to obtain the Ca2+-free sample. The harvested cells were suspended in 20 mM Tris–HCl buffer (pH 8.0) containing 500 mM NaCl and 20 mM imidazole and were disrupted by sonication at 4 °C. After the cell debris was removed by centrifugation, the supernatant was applied to a Ni–NTA column (Qiagen) and the protein was eluted by 200 mM imidazole. The fractions containing the enzyme were pooled and the buffer was exchanged with 10 mM Tris–HCl (pH 8.0). After digestion of the His-tag using the Turbo-3C precision protease (Accelagen), the protein solution was loaded into a MonoQ Sepharose column (GE Healthcare) and eluted using a linear gradient of NaCl with the AKTA purifier system (GE Healthcare). Finally, the buffer was exchanged with 10 mM Tris–HCl (pH 8.0) and the protein concentrations were determined using a UV absorption coefficient at 280 nm of 4.02 × 104 M−1 cm−1.
CD measurements
The far-UV CD melting curves were recorded on a J-820 spectropolarimeter (JASCO). The measurements were taken for 0.04 mg mL−1 protein in 10 mM Tris–HCl (pH 8.0) containing 0, 0.25, 2.5, 25, or 250 mM CaCl2, using a quartz cell with a 1.0 cm path length. The melting curves were recorded in variable temperature mode at 222 nm, from 20 to 75, 80, or 85 °C with a heating rate of 1.0 °C min−1. The analysis of the transition curves obtained by temperature-scanning CD measurements was performed on the basis of a two-state transition model, as described previously [13, 14]. The curve fitting was carried out by the nonlinear least-squares method using the OriginPro 2017 software.
DSC measurements
DSC experiments were carried out on a VP-capillary DSC (Malvern Panalytical). The data were collected by heating the solution from 5 to 90 °C at a rate of 1 °C min−1. The sample was reheated without exchanging the solution in the cells to check the reversibility. The outer buffer solution recovered from the final dialysis experiment was used in the reference cell for each case. The protein concentrations were at 1.0 mg mL−1 in 10 mM Tris–HCl (pH 8.0) containing 0, 2.5, 25, or 250 mM CaCl2. The data were analyzed using the Origin 7.0 software supplied by the manufacturer, and the fitting analysis was performed using the Origin 5.0 software. The ΔCp was calculated from the difference in the Cp values between the unfolded and folded states at the denaturation temperature (Td). The calorimetric enthalpy change (ΔHcal) was calculated by integrating the area in each heat capacity curve, as described previously [13, 14].
MD simulations
Two structures of Cut190*S176A with and without Ca2+ were built based on the Ca2+-bound structure (PDB ID; 5zno, Fig. 1) and on the Ca2+-free one (PDB ID; 4wfi). The Ca2+-bound structure has two chains, A and B. Here, chain B was chosen, because this chain was determined with a longer sequence than chain A. Ser176 in the 4wfi structure (Ca2+-free structure) was mutated to Ala by removing the Oγ atom. In addition, Arg228 was mutated to Ser by changing the side-chain atoms, resulting in both the Ca2+-bound and the Ca2+-free structures having the same amino acid sequence. To simulate Ca2+ dependence, four computational systems of 0, 25, 100, and 250 mM CaCl2 solutions were constructed for each Cut190*S176A conformation. Here, 0, 7, 21, and 47 Ca2+ ions were soaked into the systems of 0, 25, 100, and 250 mM CaCl2, respectively, and the total charge was neutralized by adding chloride ions. For the Ca2+-bound structure, the locations of the initial Ca2+ ions were kept when soaking the ions into the systems, while the newly added ions (all ions in the case of the Ca2+-free system) were placed randomly in the solvent of the simulation box. All of the charged residues (Asp, Glu, Arg, and Lys) were treated in their charged forms. Only for the systems of 0 mM CaCl2, five sodium ions were soaked to neutralize them. Explicit water molecules were placed around the protein with a margin of 0.9 nm using a truncated dodecahedron box, with an edge length of ~ 7.43 nm. The force field parameters for the protein and water molecule were Amber ff99SB-ILDN [15] and TIP3P [16], respectively, and those of the monovalent and divalent ions were based on previous reports [17, 18].
Gromacs 2016.4 was used to prepare the systems and perform the MD simulations with a time step of 2 fs [19]. Electrostatic interactions were treated by the zero-dipole summation method [21], which performs fast calculations at high accuracy comparable to the particle mesh Ewald method [22]. The zero-dipole method has been applied to MD simulations of various systems [20, 23,24,25,26,27]. After energy minimization, ten parallel simulations were executed with each a different random seed for the initial velocities of the atoms to improve the statistics for each system. Here, first NVT (100 ps at 300 K) and NPT (100 ps at 1 bar and 300 K) equilibration simulations were executed on each of the seeds, followed by a 100-ps NVT simulation at 300 K, with position restraints applied on the solute heavy atoms during the equilibration simulations. Finally, productive runs, which amount to 8 µs of simulation in total (= 100 ns × 10 parallel simulations × 2 structures × 4 Ca2+ concentrations), were executed. The structure trajectories were stored at every 10 ps. To analyze the trajectories, the Q value, which corresponds to the atomic contact fraction with respect to each initial structure, was calculated. Here, the Q value is equal to 1 when all of the contacts are conserved and is to 0 when they are completely broken. The Q value was averaged from 30 to 100 ns over all parallel trajectories in each system. Furthermore, the Ca2+-bound system at various calcium concentrations was further analyzed by their root mean square deviation (RMSD) and by using principal component analysis (PCA).
Results
Thermal transitions of purified Cut190*S176A in the absence or presence of Ca2+ were monitored by measuring the intensity change in the CD values at 222 nm. The apparent fractions in the unfolded states were plotted as a function of temperature (Fig. 2a). The crystal structure analysis shows that the S226P mutant of Cut190 has a 35.9% α-helical and a 21.2% β-sheet structure (Fig. 1), which would reflect the process of thermal unfolding for the overall structures. The thermal unfolding was irreversible, and the CD values in the denatured state under high CaCl2 concentration were close to 0. The results indicate that the protein forms aggregation after thermal denaturation, and most of them would be precipitated, especially at high CaCl2 concentrations. The thermal denaturation profiles at high CaCl2 concentrations were different from those at low CaCl2 concentrations (Fig. 2a), suggesting that Ca2+ binding to Cut190 could change its structural properties such as protein flexibility, which closely correlates with its function. Assuming a two-state transition, the transition temperature (Tm) and the van’t Hoff enthalpy change (ΔHvH) were determined by a fitting procedure (Table 1). The Tm values were almost independent of the protein concentration (0.02, 0.04, and 0.08 mg mL−1, as analyzed in our CD study), while dependent on the heating rate (0.5, 1.0, and 2.0 °C min−1, data not shown). A higher heating rate resulted in a higher Tm value, as is typical for irreversible thermal unfolding. Thus, the Ca2+-binding effects on the thermal stability of Cut190*S176A should be comparable under the same heating rate. Similar to our previous results of Cut190 and its mutants analyzed using CD and differential scanning fluorimetry [8, 11], Ca2+ binding contributes to increase the stability of Cut190*S176A.

Thermal stability analysis. a Thermal denaturation curves of Cut190*S176A under different concentrations of CaCl2, obtained by monitoring the CD value at 222 nm. b Heat capacity curves of Cut190*S176A under different concentrations of CaCl2. The solid, dotted, broken, and thin lines indicate the DSC curves under the conditions of 0, 2.5, 25, and 250 mM CaCl2, respectively. c, d Heat capacity curves of Cut190*S176A at the first (solid line) and second (broken line) scans under the conditions of 0 (c) and 250 (d) mM CaCl2
To determine the thermodynamic parameters in further detail, the thermal stabilities of Cut190*S176A under various concentrations of CaCl2 at 0, 2.5, 25, and 250 mM were analyzed using DSC (Fig. 2b). The DSC experiments showed that each heat capacity curve had a single peak and the thermal unfolding process was typically irreversible under not only high Ca2+ concentrations but also low Ca2+ concentrations (Fig. 2c, d). The DSC data were analyzed using a model of two-state transition, and the results are summarized in Table 2. Both ΔHcal and ΔHvH were obtained at Td. The Td, ΔHcal, and ΔHvH values of Cut190*S176A in the absence of Ca2+ are 54.4 °C, 861 kJ mol−1, and 575 kJ mol−1, respectively. The stabilities increase with increasing Ca2+ concentration, similar to those analyzed using CD (Table 1). The ratio of ΔHcal/ΔHvH is clearly different from either 1 or 2, indicating that cooperative transition occurs.
To analyze the mechanism of increased stability of Cut190*S176A, MD simulations at 300 K were carried out on both the Ca2+-bound and Ca2+-free structures under various Ca2+ concentrations. For each system, the average fraction of native contacts, Q, with respect to their initial structure was calculated to investigate the relative stability. As shown in Fig. 3a, all of the average Q values exhibit near unity, meaning that the structures fluctuated around their initial ones. Comparing between the Ca2+-bound and Ca2+-free states, the values of the former are higher than those of the latter. Interestingly, the Ca2+-bound structure is sensitive to the Ca2+ concentration, where the average Q value of the Ca2+-bound structure is the lowest at 0 mM Ca2+ and the highest at 25 mM Ca2+ concentration. This result suggests that Ca2+ ions in the Ca2+ binding sites specifically act on the enzyme around 25 mM Ca2+ concentration. Additionally, higher Ca2+ concentrations result in decreasing the stability slightly, suggesting that once the Ca2+ binding sites have been fully saturated, additional Ca2+ ions start to perturb the enzyme structure. To analyze the conformational space of the Ca2+-bound state, a histogram of the RMSDs of the structures at each Ca2+ concentration with respect to their average (30–100 ns) for each concentration is plotted in Fig. 3b. The distribution for 25 mM Ca2+ is the narrowest with the highest peak, and furthermore, the location of the peak along the RMSD axis is the lowest. This suggests a relatively small conformational variability in the ensemble and indicates that the protein at this concentration is the most stable. The remaining concentrations have their respective peaks at similar heights and widths, but the location of the peak along the RMSD axis varies between these concentrations. Here, 0 mM has its peak at the highest RMSD level, suggesting that at 0 mM the structures sample a wide conformational space, indicating it is the least stable system. This result is also collaborated by the PCA plots in Fig. 3c–g, where the 25 mM Ca2+ simulations sampled in a relatively small conformational space, while the 0 and 100 mM simulations sampled in a larger conformational space with 250 mM in between. Finally, Fig. 3h shows representative structures taken from each of the ensembles and some flexibility in the loop regions near the Ca2+ ions (Ser75-Gly83, Pro200-Pro213, [Glu220-His230, Asp250-Ala255, Leu286-Glu296]) can be seen, suggesting higher Ca2+ concentrations could interfere with the bound Ca2+ ions and thus disrupt protein stability.


Dependence of the Ca2+ concentration on the thermal stability obtained from the MD simulations. a Fraction of native contacts (Q-value) comparing the relative stability of the protein for the Ca2+-bound and Ca2+-free systems at four concentrations (averages with error bars), where white, red, green, and blue correspond to 0, 25, 100, and 250 mM. b Histogram of RMSDs of Ca2+-bound structures for each concentrations against their respective average structures, where black, red, green, and blue correspond to 0, 25, 100, and 250 mM. c Trajectories projected onto the first principal component (PC1) and the second one (PC2) obtained from PCA of the heavy atoms of the Ca2+-bound structures where black, red, green, and blue correspond to 0, 25, 100, and 250 mM, and X corresponds to the X-ray structure (Fig. 1). d–g Potential of mean force (PMF) for 0, 25, 100, and 250 mM, respectively, projected on to the PCA space from Fig. 3c. h Plot of representative structures at each concentration drawn using Molmil [32], where white, black, red, green, and blue are the X-ray, 0, 25, 100, and 250 mM structures, respectively. (Color figure online)
Discussion
In this study, Ca2+-dependent folding thermodynamics of Cut190*S176A were evaluated by using CD, DSC and MD simulations. Both CD and DSC results show that the stability increases with increasing CaCl2 concentration. The present results of Tm values are comparable to those of Cut190 mutants with an N-terminal His-tag, reported previously [8, 11]. In order to analyze the thermodynamic origin of the stability differences, the thermodynamic parameters at 53.6 °C, the denaturation temperature of Cut190*S176A in the Ca2+-free state, were calculated using the ΔCp values at the respective conditions, as described previously [13, 14]. Within the narrow temperature range around the Td values, the error of the calculated ΔG and ΔH values at the reference temperature should be small, even if the ΔCp values obtained contain errors. The calculated parameters are summarized in Table 3. The increased thermal stability induced upon the Ca2+ binding is mainly due to the favorable ΔH, which is partially compensated by an unfavorable ΔS. The favorable ΔH could be explained by the increased contact ratios in the Ca2+-bound state shown by the MD simulations. The increased contacts would result in the decreased fluctuation of Cut190*S176A in the Ca2+-bound state, reflecting to the unfavorable ΔS.
Figure 4 shows a plot of ln[CaCl2] versus T −1m and T −1d determined from CD and DSC, respectively. When the Ca2+ concentration in the free state is higher than that in the bound state, the plot corresponds to the van’t Hoff analysis, whose slope provides ΔHvH. As reported previously [28, 29], when the ligand concentration is much higher than the dissociation constant (KD) for the ligand–protein interaction, the protein concentration in the bound state is much larger than that in the free state, generating a single DSC peak derived from the ligand-bound protein. Preliminary isothermal titration calorimetry experiments showed that the KD value for Ca2+ binding to Cut190*S176A was in the mM order (unpublished results). The Ca2+ concentration range analyzed in this study was between 0.25 and 250 mM. At the lower Ca2+ concentrations, most of the enzymes should exist in the Ca2+-free state. However, the DSC peak shifts to a higher temperature with little change in its shape, and the plot in Fig. 4 could be fitted linearly, providing ΔHvH values to be 598 and 546 kJ mol−1, which are comparable to those determined from CD and DSC (Tables 1 and 2). One possibility to explain this phenomena is that the association and dissociation rates of Ca2+ binding are so fast, mainly due to the electrostatic interaction [30, 31], that Cut190*S176A including the state close to thermal denaturation could bind to Ca2+, resulting in an increased denaturation temperature by forming a transient complex.

Dependence of the Ca2+ concentration on the thermal stability. The CaCl2 concentrations are plotted against T −1m from CD (◇) or T −1d from DSC (◯)
MD simulations show that the enzyme structure does not significantly change upon different Ca2+ concentrations. Especially in the Ca2+-free structure, no effect of the Ca2+ concentrations was observed. In the Ca2+-bound structure, the Ca2+ ions perturb local regions based on the Q value, RMSD, and the PCA analyses. The mechanism is suggested as follows. In the three models with Ca2+ ions included (25, 100, and 250 mM), three Ca2+ ions are in the binding sites forming stable loop structures (Ser75-Gly83, Pro200-Pro213, [Glu220-His230, Asp250-Ala255, Leu286-Glu296]). These bound Ca2+ ions also interact with the surrounding environment. When the Ca2+ concentration is high (e.g., 250 mM), the bound ions presumably frequently interact with other Ca2+ ions and/or counter chloride ions. As a result, the loop structures along with the bound ions tend to fluctuate. Therefore, the Ca2+ concentration of 25 mM is optimal to stabilize the Ca2+-bound structure. It seems to be difficult to estimate how much the Ca2+ concentration in the MD simulations corresponds to that in the solution in the experimental condition, because the box size of the MD system is limited. The Ca2+ in the MD simulations might have additional effects in solution, similar to the environments at higher Ca2+ concentrations. The structure transition mechanism from the Ca2+-free to the Ca2+-bound structures, however, remains unclear. Even at high Ca2+ concentrations, no conformational change in the Ca2+-free structure to the conformation of the Ca2+-bound one was observed, although the Ca2+ ions reached equilibrium. This suggests that the Ca2+ ions need to bind for a considerable amount of time before the conformational change will occur. Thus, although this work has clarified the effect of Ca2+ ions on the Ca2+-bound structure, further long MD simulations (e.g., > 1 ms) are required to show this transition, which at this point in time are unfeasible. Alternatively, enhanced sampling simulations such as multicanonical MD could be performed to speed up this process [21, 26], but such an approach is beyond the scope of this work.
Conclusions
The present MD simulations of Cut190*S176A in the absence or presence of Ca2+ are in good correlation with the experimental results of folding thermodynamics that Ca2+ binding to Cut190*S176A increases the thermal stability by increasing ΔH and decreasing ΔS, probably due to the increase in intramolecular interactions and the decrease in protein flexibility. The Ca2+ binding to the three sites of Cut190*S176A would change its structure into the more stable state with increasing intramolecular interactions.
References
Purdy RE, Kolattukudy PE. Hydrolysis of plant cuticle by plant pathogens. Properties of cutinase I, cutinase II, and a nonspecific esterase isolated from Fusarium solani pisi. Biochemistry. 1975;14:2832–40.
Nyyssölä A. Which properties of cutinases are important for applications? Appl Microbiol Biotechnol. 2015;99:4931–42.
Carvalho CM, Aires-Barros MR, Cabral JM. Cutinase: from molecular level to bioprocess development. Biotechnol Bioeng. 1999;66:17–34.
Bornscheuer UT. Microbial carboxyl esterases: classification, properties and applications in biocatalysis. FEMS Microbiol Rev. 2002;26:73–81.
Pio TF, Macedo GA. Cutinases: properties and industrial applications. Adv Appl Microbiol. 2009;66:77–95.
Zimmermann W, Billig S. Enzymes for the biofunctionalization of poly(ethylene terephthalate). Adv Biochem Eng Biotechnol. 2010;125:97–120.
Chen S, Su L, Chen J, Wu J. Cutinases: characteristics, preparation, and application. Biotechnol Adv. 2013;31:1754–67.
Kawai F, Oda M, Tamashiro T, Waku T, Tanaka N, Yamamoto M, Mizushima H, Miyakawa T, Tanokura M. A novel Ca2+-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl Microbiol Biotechnol. 2014;98:10053–64.
Hu X, Thumarat U, Zhang X, Tang M, Kawai F. Diversity of polyester-degrading bacteria in compost and molecular analysis of a thermoactive esterase from Thermobifida alba AHK119. Appl Microbiol Biotechnol. 2010;87:771–9.
Thumarat U, Kawabata T, Nakajima M, Nakajima H, Sugiyama A, Yazaki K, Tada T, Waku T, Tanaka N, Kawai F. Comparison of genetic structures and biochemical properties of tandem cutinase-type polyesterases from Thermobifida alba AHK119. J Biosci Bioeng. 2015;120:491–7.
Miyakawa T, Mizushima H, Ohtsuka J, Oda M, Kawai F, Tanokura M. Structural basis for the Ca2+-enhanced thermostability and activity of PET-degrading cutinase-like enzyme from Saccharomonospora viridis AHK190. Appl Microbiol Biotechnol. 2015;99:4297–307.
Miller JH. Experiments in molecular biology. New York: Cold Spring Harbor Laboratory; 1972. p. 431–3.
Inaba S, Fukada H, Ikegami T, Oda M. Thermodynamic effects of multiple protein conformations on stability and DNA binding. Arch Biochem Biophys. 2013;537:225–32.
Inaba S, Fukada H, Oda M. Effect of a salt-bridge between inter-repeats on the 3D structure of the c-Myb DNA-binding domain revealed by thermodynamic analysis. J Therm Anal Calorim. 2018;131:335–41.
Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–8.
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35.
Joung IS, Cheatham TE 3rd. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020–41.
Li P, Roberts BP, Chakravorty DK, Merz KM Jr. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J Chem Theory Comput. 2013;9:2733–48.
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25.
Fukuda I, Kamiya N, Yonezawa Y, Nakamura H. Simple and accurate scheme to compute electrostatic interaction: zero-dipole summation technique for molecular system and application to bulk water. J Chem Phys. 2012;137:054314.
Bekker G-J, Kamiya N, Araki M, Fukuda I, Okuno Y, Nakamura H. Accurate prediction of complex structure and affinity for a flexible protein receptor and its inhibitor. J Chem Theory Comput. 2017;13:2389–99.
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93.
Kamiya N, Fukuda I, Nakamura H. Application of zero-dipole summation method to molecular dynamics simulations of a membrane protein system Chem. Phys Lett. 2013;568–569:26–32.
Arakawa T, Kamiya N, Nakamura H, Fukuda I. Molecular dynamics simulations of double-stranded DNA in an explicit solvent model with the zero-dipole summation method. PLoS ONE. 2013;8:e76606.
Kamiya N, Mashimo T, Takano Y, Kon T, Kurisu G, Nakamura H. Elastic properties of dynein motor domain obtained from all-atom molecular dynamics simulations. Protein Eng Des Sel. 2016;29:317–25.
Nishigami H, Kamiya N, Nakamura H. Revisiting antibody modeling assessment for CDR-H3 loop. Protein Eng Des Sel. 2016;29:477–84.
Oda M, Inaba S, Kamiya N, Bekker G-J, Mikami B. Structural and thermodynamic characterization of endo-1,3-β-glucanase: insights into the substrate recognition mechanism. Biochim Biophys Acta. 2018;1866:415–25.
Fukada H, Sturtevant JM, Quiocho FA. Thermodynamics of the binding of L-arabinose and of D-galactose to the L-arabinose-binding protein of Escherichia coli. J Biol Chem. 1983;258:13193–8.
Sturtevant JM. Biochemical applications of differential scanning calorimetry. Ann Rev Phys Chem. 1987;38:463–88.
Wade RC, Gabdoulline RR, Lüdemann SK, Lounnas V. Proc Natl Acad Sci USA. 1998;95:5942–9.
Oda M, Nakamura H. Thermodynamic and kinetic analyses for understanding sequence-specific DNA recognition. Genes Cells. 2000;5:319–26.
Bekker G-J, Nakamura H, Kinjo AR. Molmil: a molecular viewer for the PDB and beyond. J Cheminform. 2016;8:42.
Acknowledgements
The authors thank Ms. Ayako Miki and Akane Senga of Kyoto Prefectural University for protein preparation, Dr. Nobutaka Numoto of Tokyo Medical and Dental University for crystal structure information, and Dr. Harumi Fukada of Osaka Prefecture University for critical reading of the manuscript. The CD and DSC experiments were performed at the Okinawa Institute Science and Technology Graduate University (OIST). This work was supported by the Research Grant from Institute for Fermentation, Osaka (IFO) to M.O. This work was also supported by Grand-in-Aid for Scientific Research C (JP16K07331) from the Japan Society for the Promotion of Science (JSPS) to N.K. It was performed in part under the Cooperative Research Program of the Institute for Protein Research, Osaka University, CR-17-05 and CR-18-02. Computational resources of a PC cluster were partly provided by the HPCI Research Project (hp150146 and hp170024).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Inaba, S., Kamiya, N., Bekker, GJ. et al. Folding thermodynamics of PET-hydrolyzing enzyme Cut190 depending on Ca2+ concentration. J Therm Anal Calorim 135, 2655–2663 (2019). https://doi.org/10.1007/s10973-018-7447-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7447-9