
Abstract
Palladium(II) complexes with tetradentate Schiff bases, Pd(acacpda)(I) and Pd(acacdmpda)(II) (acacpda2−—N,N′-(propylene)-bis(acetylacetoneiminato-), acacdmpda2−—N,N′-(2,2-dimethylpropylene)-bis(acetylacetoneiminato-)), have been synthesized, purified and characterized by elemental analysis, IR and NMR spectroscopy. Complex I was obtained for the first time, and its solid-state structure was investigated by single-crystal X-ray diffraction. It has a discrete molecular structure with the Pd2+ ion being in a distorted square-planar coordination environment formed by N and O atoms of the acacpda2− ligand. The thermal properties of complexes I and II in condensed phase were investigated by thermogravimetry. Compound II transfers into the gas phase almost completely, while compound I evaporates with a partial decomposition. The temperature dependences of saturated vapor pressure were measured by the flow method in the temperature intervals (423–457) and (422–478) K for I and II, respectively. Both complexes were shown to exhibit an excellent stability upon sublimation. The thermodynamic parameters of sublimation processes were calculated to be: ΔsubH440 = (133 ± 4) kJ mol−1, ΔsubS 0440 = (210 ± 9) J (mol K)−1 for I, ΔsubH450 = (135 ± 1) kJ mol−1, ΔsubS 0450 = (218 ± 3) J (mol K)−1 for II. It has been found that the complex II with a more bulky Schiff base ligand possesses slightly higher vapor pressure values as compared to I, which is probably due to the lower contribution of weak intermolecular interactions occurring in the crystal structure of II.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nowadays, bimetallic films and nanoparticles based on palladium are widely used in different fields of application, for example, as materials for magnetooptical recording devices [1, 2], biosensors for methotrexate detection [3], water purification and decolorization systems [4, 5], hydrogen purification membranes [6,7,8], hydrogen sensors [9, 10] and in catalysis of a number of chemical reactions such as oxidation, reduction, hydrogenation, cross-coupling and others [10,11,12,13,14,15].
The perspective techniques to obtain such bimetallic structures are metal organic chemical vapor deposition (MOCVD) and related methods such as atomic layer deposition (ALD), etc. The main advantages of these methods are complete preside control over the properties (composition and microstructure/size) of the produced films and nanoparticles by means of variation a number of deposition parameters as well as the possibility of formation of target structures on the complicated shape objects and the absence of side components (for example, protecting agents in the case of the colloidal synthesis methods). The investigation of the formation of Pd-based bimetallic systems by chemical vapor-phase deposition techniques has been developing ever more actively since 1990s: various structures have been obtained with a number of metals including silver [16, 17], platinum [18,19,20,21], ruthenium [21], gold [22], copper [23] and nickel [24,25,26]. However, it should be noted that in order to realize all the advantages of the MOCVD and related methods, the volatile precursors of both components apart from the standard requirements for thermal properties (sufficient volatility, thermal stability and others [27]) should possess their experimental compatibility. Thus, in order to ensure further progress in the area discussed, it is necessary to expand a number of thermochemically characterized volatile palladium complexes to select the optimal combinations of the precursors for effective deposition of each bimetallic system.
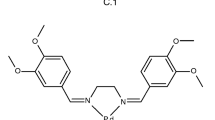
To date, palladium complexes with the following ligands have been proposed as volatile MOCVD/ALD precursors: β-diketonates, β-iminoketonates, β-enaminodiones, dialkyldithiocarbamates, dialkyldithiophosphates, cyclopentadienyls, allyls, alkenols, etc. [28,29,30,31,32,33,34,35,36]. This work is devoted to a new class of palladium MOCVD precursors—Pd(II) complexes with tetradentate Schiff bases (Fig. 1). Preliminary research has shown that the replacement of the ethylene diimine bridge in the ligand with propylene one increases the volatility of the formed complexes [37]. So, the objects of the present investigation are palladium complexes with propylene-diimine bridge containing Schiff bases: Pd(acacpda) I and Pd(acacdmpda) II (Fig. 1, acacpda2−—N,N′-(propylene)-bis(acetylacetoneiminato-), acacdmpda2−—N,N′-(2,2-dimethylpropylene)-bis(acetylacetoneiminato-)). The paper presents the synthesis, detailed characterization and thermochemical investigation of the complexes including the crystal structure of compound I obtained for the first time.

General structure formula of palladium(II) complexes with tetradentate Schiff bases studied
Experimental
Materials and methods
The following reagents were used in the presented work: palladium chloride (Krastsvetmet, palladium content 59.845%), acetylacetone (Acros Organics, purity > 99%), propylenediamine (Merck, purity > 99%), 2,2-dimethylpropylenediamine (Aldrich, purity > 99%). Acetonitrile (Kriokhrom, purity > 99%), hexane (“Reaktiv”, purity > 98%), toluene (“Reaktiv” , purity > 98%) were used as a solvents. All of the reactants were used as supplied.
The elemental analysis was carried out on a CHNS-analyzer vario MICRO cube with the accuracy 0.5%. IR spectra were recorded on a Scimitar FTS 2000 spectrometer in the 400–4000 cm−1 range; the samples were prepared by pressing the pellets with KBr. NMR spectra were recorded on an Avance 500 spectrometer at 500 MHz for 1H NMR and 125 MHz for 13C NMR. The primary spectral data are presented in Supplementary Materials. The melting points (m.p.) were determined by the visual method using the Kofler bench.
Synthesis of Schiff bases and palladium complexes
H2acacpda was synthesized according to the method [38] through the interaction of 2.15 mL (21.0 mmol) acetylacetone and 0.90 mL (10.5 mmol) propylenediamine by means of stirring under reflux in the absence of a solvent. The product was purified by recrystallization from hexane, yield 80%. Anal. Calcd. for C13H22N2O2 (mass. %): C 65.5, H 9.3, N 11.8. Found C 65.2, H 9.2, N 11.7. IR spectrum (cm−1): 726, 751, 1026, 1088, 1138, 1282, 1353, 1440, 1514, 1568, 1607, 2875, 2953, 3547.
H2acacdmpda was synthesized and purified similarly to H2acacpda from 1.95 mL (18.8 mmol) of Hacac and 1.20 mL (9.41 mmol) of 2,2-dimethylpropylenediamine. Yield 80%. Anal. Calcd. for C15H26N2O2: C 67.6, H 9.8, N 10.5. Found: C 67.7, H 9.7, N 10.7. IR spectrum (cm−1): 751, 807, 975, 1023, 1104, 1197, 1290, 1359, 1442, 1521, 1572, 1611, 2880, 2970, 3388.
Pd(acacpda)(I) was obtained according to modified procedure [39]. Firstly, Pd(CH3CN)2Cl2 complex was prepared by dissolving of 0.518 g (2.92 mmol) PdCl2 in 70 mL of acetonitrile at slight heating and then the 20 mL of acetonitrile solution of H2acacpda (0.696 g, 2.92 mmol) was added to reaction mixture which was further kept for 30 min. Then, the NaOH aqueous solution (0.234 g, 5.85 mmol) was added. The product was separated as a precipitate after 30 min of stirring at room temperature by adding of 500 mL of water. An additional amount of the product was obtained by toluene extraction of the filtrate. The complex was purified by vacuum sublimation (443 K, 6.67 Pa). Yield 62% (0.615 g). m. p. 468 K. Anal. Calcd. for PdC13H20N2O2(I): C 45.6, H 5.9, N 8.2. Found: C 45.4, H 6.0, N 8.1. IR spectrum (cm−1): 423, 460, 758, 952, 1015, 1119, 1252, 1274, 1359, 1405, 1466, 1512, 1577, 2913, 2938. 1H NMR (CDCl3, 7.26 ppm): 1.9564 (s, 6H, CH3C=N), 1.9767 (s, 6H, CH3C=O), 2.0532 (m, 2H, (CH2)CH2(CH2)), 3.0389 (t, 4H, (CH2)CH2(CH2)), 4.8548 (s, 2H, CH). 13C NMR (CDCl3, 77.2 ppm): 23.0504 (CH3C=N), 24.5162 (CH3C=O), 32.4997 (CH2)CH2(CH2), 51.2816 (CH), 99.4074 (CH2–N), 164.3894 (C=N), 176.7554 (C=O).
Pd(acacdmpda)(II) was synthesized in a similar way to I from 0.333 g (1.88 mmol) of PdCl2, 0.500 g (1.88 mmol) of H2acacdmpda, and 0.180 g (4.50 mmol) of NaOH. The complex was purified by vacuum sublimation ((433–443) K, 6.67 Pa). Yield 88% (0.543 g). m. p. 487 K. Anal. Calcd. for PdC15H24N2O2(II): C 48.4, H 6.5, N 7.6. Found: C 48.0, H 6.5, N 7.6. IR spectrum (cm−1): 449, 469, 756, 952, 1013, 1112, 1232, 1273, 1332, 1365, 1398, 1456, 1511, 1571, 2916, 2956. 1H NMR (CDCl3, 7.26 ppm): 1.1141 (s, 6H, C(CH3)2), 1.9522 (s, 6H, CH3C=N), 1.9899 (s, 6H, CH3C=O), 3.0442 (s, 4H, CH2C–N), 4.8332 (s, 2H, CH). 13C NMR (CDCl3, 77.2 ppm): 22.4577 (CH3C=N), 24.7277 (CH3)2C(CH2)2, 25.9650 (CH3C=O), 36.5668 (CH3)2C(CH2)2, 61.1032 (CH), 99.6479 (CH2–N), 163.3507 (C=N), 176.6955 (C=O).
Single-crystal X-ray diffraction
A yellow needle crystal of I was selected under a microscope and then mounted to the tip of the thin glass fiber with epoxy resin. X-ray intensity data were collected on a Bruker-Nonius X8 Apex CCD diffractometer at 296(2) K using graphite monochromatized MoKα radiation (λ = 0.71,073 Å). The standard technique was used (combined φ and ω scans of narrow frames). Data reduction and multi-scan absorption were carried out using the SADABS [40]. The structure was solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXTL software package [40]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of acacpda2− ligand were placed in geometrically calculated positions and refined as riding on their parent carbon atoms with Uiso(H) = 1.5Ueq(C) for methyl hydrogen atoms and Uiso(H) = 1.2Ueq(C) for methylene and β-hydrogen atoms. The molecular graphics were performed using DIAMOND program [41]. Crystallographic data and selected refinement details are given in Table 1. The main bond lengths and angles are presented in Table 2. The atomic positional and thermal parameters, full lists of bond lengths and angles were deposited at the Cambridge Crystallographic Data Centre under the reference number CCDC 1576778 and are available free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Thermal investigation
The thermogravitmetric investigation (TG/DTA) was performed on a Netzsch TG 209 F1 Iris thermal analyzer in an open Al2O3 crucible in a helium flow (30.0 mL min−1). The heating rate was 10 K min−1; sample mass was (10 ± 1) mg.
The temperature dependencies of saturated vapor pressure of the complexes were measured by flow (transpiration) method in the atmosphere of dry inert gas carrier (helium) using the equipment presented in Fig. 2. The principles of the method and the experimental procedure used are described in [42, 43] and [44], respectively. During the experiment, the substance in source 1 is transferred to the gas phase by heating and carried by the carrier gas flow through the porous plugs 5 to the receiver 2 where it condenses on the cold walls. The amounts of substance in source and receiver were determined gravimetrically (uncertainty is 5 × 10−4 g). The sublimation is performed by a calibrated heater 6 with a thermocouple sensor 7. The temperature was measured by thermal control device 8 with uncertainty 0.5 K, the carrier gas flow was measured by flow mass (11, 12), and helium flow rate was measured by device 13 with uncertainty 2%. The total relative standard uncertainty of this method was not above 5%. The measurements were taken in the quasi-equilibrium conditions, which were proved by the independence of the vapor pressure on the helium flow rate. The partial pressure of saturated vapor over complexes was calculated according to Eq. 1:
Ptotal total pressure in a system; n number of moles of carried compound; N number of moles of helium. The calculations were based on the assumption that the substance vaporizes in the monomolecular form. The experimental results were processed statistically using the objective function recommended in [45]. Data obtained are presented in Table 1S (Supplementary Materials).

Experimental setup for tensimetric measurements by flow method: 1—source, 2—receiver, 3—analyzed substance in source, 4—condensed substance in receiver, 5—porous plugs, 6—heater, 7—thermocouple sensors, 8—thermal control device, 9—carrier gas cylinder, 10—gas drying unit, 11, 12—gas consumption regulator (flow mass), 13—carrier gas flow rate measuring device
Results and discussion
Synthesis and characterization
Palladium complexes I and II were synthesized by the interaction of Pd(CH3CN)2Cl2 prepared in situ by dissolving of PdCl2 in CH3CN under reflux with corresponding Schiff base in acetonitrile and an aqueous solution of alkali (Eq. 2). The products were precipitated by adding a large amount of water. The complexes I and II are yellow crystalline powders stable on storage in air at reduced temperature for at least 6 months.
The comparison of IR spectra of the complexes with IR spectra of corresponded Schiff bases is presented in Table 3. The presence of stretching vibrational bands ν(Pd–N), ν(Pd–O) in the IR spectra of the complexes as well as the absence of bands ν(N–H), ν(O–H) indicates the complexation. Shifts of absorption bands ν(C=O), ν(C=N) toward lower frequencies also confirm the formation of the complexes.
The expected signal positions in the 1H NMR and 13C NMR spectra, as well as the integrated intensities of the 1H NMR spectra signals of the complexes, also confirm their structure in solution (Fig. 1). The proton signals of the CH3C=N, CH3C=O groups are located closely, with a larger shift of CH3C=O proton group toward the stronger field. The signals of the protons of other aliphatic fragments are easily recognizable and typical. Comparison of the 1H NMR spectra of complexes I and II with the spectra of corresponding Schiff bases [45] shows the absence of acidic proton signal at about 11 ppm, indicating the complexation.
According to single-crystal X-ray diffraction analysis, the complex I has a molecular structure consisting of discrete Pd(acacpda) molecules (Fig. 3), which can be best compared with the structure of the related complex Pd(acacdmpda), II [37]. All atoms of the complex molecule are crystallographycally independent and occupy general positions. Within the molecule, the Pd2+ ion has a distorted square-planar coordination environment formed by imino N and carbonyl O atoms of the tetradentate acacpda2− ligand. The bond lengths and angles around the palladium center are, as expected, close to the corresponding values found in II (Table 2). In particular, the average values for Pd–O and Pd–N bond lengths are 1.995(1) and 2.023(1) Å, respectively, while the chelate N–Pd–O angles are 91.98(14) and 92.13(13)°. The N–Pd–N and O–Pd–O angles are 96.08(14) and 79.73(13)°, respectively. Due to the tetradentate coordination of the acacpda2− ligand, the Pd(acacpda) molecule contains two conjugated six-membered metallocycles (PdOCCCN) and one six-membered metallocycle (PdNCCCN). Both (PdOCCCN) metallocycles are rather nonplanar, with the angles of folding along the O(1)N(1) and O(2)N(2) lines being 17.1 and 20.1°, respectively. The (PdNCCCN) metallocycle contains no conjugated bonds and deviates considerably from planarity; its geometry is characterized by the angles of folding along the C(6)C(8) and N(1)N(2) lines, which are 63.0° and 17.9°, respectively. Therefore, the (PdNCCCN) metallocycle can be best described as having a distorted boat conformation with the palladium atom at the front and the opposing carbon atom at the back.

Thermal ellipsoid representation of I with the atomic numbering scheme (ellipsoids are drawn at the 50% probability level)
At a supramolecular level, both complexes I and II exhibit C–H···O hydrogen bonds, leading to a chain-like arrangement of molecules in the crystals (Fig. 4). Despite the weakness of these interactions, their relative strength can be compared based on the C···O distances. In I, the central (α) hydrogen atom of the β-diketone unit participates in the formation of hydrogen bond, showing the C···O distance of 3.32 Å, while in II, the methylene hydrogen atoms make weaker hydrogen bonds with the C···O distances of 3.48 and 3.51 Å. Moreover, the hydrogen-bonded chains of Pd(acacpda) and Pd(acacdmpda) molecules are organized in different ways and, as one can expect, their packing motifs and densities are rather different. In particular, the chains of Pd(acacpda) molecules in I have a zigzag shape and are characterized by the following Pd···Pd distances: 7.89 Å between the adjacent Pd(acacpda) molecules within the chain and 6.40 Å between the Pd(acacpda) molecules from the adjacent chains. In II, the chains of Pd(acacdmpda) molecules have a more linear shape with the corresponding Pd···Pd distances of 6.18 and 8.05 Å [37]. Most likely, the reason for the differences in hydrogen-bonding patterns in I and II lies in the higher bulkiness of acacdmpda2− ligand as compared to that of acacpda2− ligand. The presence of two methyl groups in propylene-diimine bridge of acacdmpda2− ligand results in a less dense packing of the Pd(acacdmpda) molecules in the crystal of II.

Packing diagram of I viewed along the a axis. H atoms are omitted for clarity. Hydrogen bonds C–H···O are shown as dashed lines
Thermal properties
Thermal properties of the complexes in condensed phase were investigated by TG/DTA. The endoeffects on the DTA curves at 467 K and 485 K (onset temperatures) for I and II, respectively, correspond to the melting process, that have been confirmed visually on the Kofler’s table. The mass loss curves are presented in Fig. 5. Under the conditions of the TG experiments, the complexes evaporate in the temperature range (498–598) K. Compound II transfers into gas phase almost quantitatively (mass loss 98.0%), while compound I evaporates with a partial decomposition (mass loss 87.3%). Temperatures of 50% mass loss (T50%) are close (576 and 573 K for I and II, respectively); consequently, the complexes appear to be comparable in volatility.

Mass loss curves of complexes Pd(acacpda)(I) and Pd(acacdmpda)(II)
In quasi-equilibrium conditions of tensimetric experiments, in the temperature intervals (423–457) K and (422–478) K for I and II, respectively, the difference in volatility of the complexes was more accurately revealed. The mass of substance in receiver was equal to the mass of evaporated one in the all investigated temperature range, indicating the stability of the complexes during sublimation processes. The results obtained are presented in Fig. 6 and Table 1S (Supplementary Materials). The thermodynamic parameters of sublimation and logarithmic temperature dependences of the saturated vapor pressure are listed in Table 4. It should be noted that the differences in experimental pressure values from the calculated ones (Table 1S) are not exceed standard uncertainties in temperature and pressure measurements, while the random character of these deviations indicates the absence of a systematic error in the measurements.

Temperature dependences of the saturated vapor pressure of complexes Pd(acacpda)(I) and Pd(acacdmpda)(II)
The data obtained allow us to conclude that the insertion of methyl substituents in the diimine bridge slightly increases the volatility of the complexes: vapor pressure values of complexes I and II at 433 K are 0.82 and 1.35 Pa, respectively. It should be noted that the same regularities were observed for analogous copper(II) complexes by means of TG method [46]. The slight difference in volatility observed for the complexes under study could be associated with the weak intermolecular interactions revealed in their crystal packing. In fact, the structure of less volatile complex I is characterized by stronger C–H···O hydrogen bonds described above. Moreover, the packing of complex I is more dense as it could be also concluded from the higher calculated density of this compound (1.631 g cm−1) in comparison with that of complex II (1.526 g cm−1), whereas the molecular weight of complex I is lower, and the number of molecules in the unit cell is the same. The loosening of the crystalline package due to methyl substituents introduced into the diimine bridge of the Schiff base ligand could also be one of the factors that lead to an increase in compound volatility.
At the same time, palladium chelates I and II are characterized by the lower volatility in comparison with palladium complexes with bidentate Schiff bases (β-iminoketonates). For example, the saturated vapor pressure of the simplest representative of palladium β-iminoketonates with alkyl substituent at the nitrogen atom, Pd(Mei-acac)2 (Mei-acac−—4-methylamino-3-penten-2-onato-), is almost 30 times higher (34.85 Pa at 433 K) [32]. The same regularity seems to be typical for complexes of 3d-transition metals with coordination polyhedrons close to the square-planar namely Cu(II), Co(II), Ni(II) [46,47,48,49,50,51]. However, prior to the present work, the quantitative tensimetric data were available only for nickel complexes (saturated vapor pressure of Ni(acacen) at 433 K is 0.83 Pa, Ni(i-acac)2—32.1 Pa (acacen2−—N,N′-(ethylene)-bis(acetylacetoneiminato-), i-acac−—4-amino-3-penten-2-onato-) [50, 51], whereas for the other complexes it was shown only qualitatively (by TG experiments).
Nevertheless, it is worth noting that the vapor pressure values and stability of complexes I and II during sublimation make it possible to use these precursors in the typical MOCVD reactors. In this aspect, the tensimetric data obtained are useful to select the temperature parameters of the evaporator (precursor vapor source) in MOCVD experiments to control the partial pressure of palladium precursor during deposition of bimetallic nanostructures.
Conclusions
Volatile palladium(II) complexes Pd(acacpda)(I) and Pd(acacdmpda)(II) with tetradentate Schiff bases containing propylene-diimine bridge were synthesized and investigated as potential MOCVD precursors. The compounds obtained were fully characterized by elemental analysis, IR and NMR spectroscopy, and solid-state structure of complex I obtained for the first time was investigated by single-crystal X-ray diffraction. The influence of the ligand structure on the complexes thermal properties (volatility and stability) were studied using thermogravimetric analysis and tensimetry (flow method). Temperature dependences of saturated vapor pressure have been measured in the temperature intervals (423–457) K and (422–478) K for I and II, respectively, and thermodynamic parameters of sublimation were calculated.
It has been shown that the introducing methyl groups to the central atom of diimine bridge in Schiff base ligand (complex II vs. complex I) does not lead to significant changes in the palladium coordination polyhedron being a distorted square-planar formed by imino N and carbonyl O atoms. However, the modification of the ligand under consideration results in noticeable changes in the crystal packing, which is reflected in the thermal properties of the complexes. In particular, complex II is characterized by slightly higher volatility than I (the difference in vapor pressure values is about 0.2 order of magnitude). This observation could be explained in terms of extenuation of weak hydrogen intermolecular interactions in the crystal structure of II with a more bulky Schiff base ligand and its lower packing density.
Although the evaporation of complex I is accompanied by a partial decomposition, the sublimation processes for both complexes have been shown to proceed purely. Consequently, the thermal stability of the complexes I and II as well as the vapor pressure values are sufficient for the use in MOCVD processes.
References
Duan S, Wang R. Bimetallic nanostructures with magnetic and noble metals and their physicochemical applications. Prog Nat Sci Mater Int. 2013;23:113–26.
Vivas LG, Figueroa AI, Bartolomé F, Rubín J, García LM, Deranlot C, Petroff F, Ruiz L, González-Calbet JM, Brookes NB, Wilhelm F. Structural and magnetic properties of granular CoPd multilayers. J Magn Magn Mater. 2016;400:248–52.
Ghadimi H, Nasiri-Tabrizi B, Nia PM, Basirun WJ, Tehrani RM, Lorestani F. Nanocomposites of nitrogen-doped graphene decorated with a palladium silver bimetallic alloy for use as a biosensor for methotrexate detection. RSC Adv. 2015;5:99555–65.
Patel R, Suresh S. Decolourization of azo dyes using magnesium–palladium system. J Hazard Mater. 2006;137:1729–41.
Chaplin BP, Reinhard M, Schneider WF, Schüth C, Shapley JR, Strathmann TJ, Werth CJ. Critical review of Pd-based catalytic treatment of priority contaminants in water. Environ Sci Technol. 2012;46:3655–70.
Yun S, Oyama ST. Correlations in palladium membranes for hydrogen separation: a review. J Membr Sci. 2011;375:28–45.
Burkhanov GS, Gorina NB, Kolchugina NB, Roshan NR, Slovetsky DI, Chistov EM. Palladium-based alloy membranes for separation of high purity hydrogen from hydrogen-containing gas mixtures. Plat Met Rev. 2011;55(1):3–12.
Li H, Caravella A, Xu HY. Recent progress in Pd-based composite membranes. J Mater Chem. 2016;4:14069–94.
Strohfeldt N, Tittl A, Giessen H. Long-term stability of capped and buffered palladium-nickel thin films and nanostructures for plasmonic hydrogen sensing applications. Opt Mater Express. 2013;3(2):194–204.
Wadell C, Nugroho FA, Lidström E, Iandolo B, Wagner JB, Langhammer C. Hysteresis-free nanoplasmonic Pd–Au alloy hydrogen sensors. Nano Lett. 2015;15(5):3563–70.
Shu JB, Grandjean BP, Neste AV, Kaliaguine S. Catalytic palladium-based membrane reactors: a review. Can J Chem Eng. 1991;69(5):1036–60.
Molnar A. Efficient, selective, and recyclable palladium catalysts in carbon−carbon coupling reactions. Chem Rev. 2011;111:2251–320.
Yu W, Porosoff MD, Chen JG. Review of Pt-based bimetallic catalysis: from model surfaces to supported catalysts. Chem Rev. 2012;112(11):5780–817.
Gao F, Goodman DW. Pd–Au bimetallic catalysts: understanding alloy effects from planar models and (supported) nanoparticles. Chem Soc Rev. 2012;41:8009–20.
Hu S, Scudiero L, Ha S. Electronic effect of Pd-transition metal bimetallic surfaces toward formic acid electrochemical oxidation. Electrochem Commun. 2014;38:107–9.
Lu SY, Lin YZ. Pd–Ag alloy films prepared by metallorganic chemical vapor deposition process. Thin Solid Films. 2000;376(1):67–72.
Hintsho N, Petrik L, Nechaev A, Titinchi S, Ndungu P. Photo-catalytic activity of titanium dioxide carbon nanotube nano-composites modified with silver and palladium nanoparticles. Appl Catal B. 2014;156:273–83.
Hierso JC, Feurer R, Poujardieu J, Kihn Y, Kalck P. Metal-organic chemical vapor deposition in a fluidized bed as a versatile method to prepare layered bimetallic nanoparticles. J Mol Catal A. 1998;135(3):321–5.
Guerrero RM, Palacios EG, Garcia JV, Hernandez VS, Guzman AS, Meneses ER. Bimetallic Pd–Pt films prepared by MOCVD. Rev Mex Fis. 2007;53(3):194–7.
Lei Y, Liu B, Lu J, Lobo-Lapidus RJ, Wu T, Feng H, Xia X, Mane AU, Libera JA, Greeley JP, Miller JT, Elam JW. Synthesis of Pt–Pd core–shell nanostructures by atomic layer deposition: application in propane oxidative dehydrogenation to propylene. Chem Mater. 2012;24:3525-18.
Lu J, Low KB, Lei Y, Libera JA, Nicholls A, Stair PC, Elam JW. Toward atomically-precise synthesis of supported bimetallic nanoparticles using atomic layer deposition. Nat Commun. 2014;5:3264.
Wang H, Wang C, Yan H, Yi H, Lu J. Precisely-controlled synthesis of Au@ Pd core–shell bimetallic catalyst via atomic layer deposition for selective oxidation of benzyl alcohol. J Catal. 2015;324:59–68.
Krisyuk VV, Shubin YV, Senocq F, Turgambaeva AE, Duguet T, Igumenov IK, Vahlas C. Chemical vapor deposition of Pd/Cu alloy films from a new single source precursor. J Cryst Growth. 2015;414:130–4.
Jun CS, Lee KH. Palladium and palladium alloy composite membranes prepared by metal-organic chemical vapor deposition method (cold-wall). J Membr Sci. 2000;176(1):121–30.
Assaud L, Monyoncho E, Pitzschel K, Allagui A, Petit M, Hanbücken M, Baranova EA, Santinacci L. 3D-nanoarchitectured Pd/Ni catalysts prepared by atomic layer deposition for the electrooxidation of formic acid. Beilstein J Nanotechnol. 2014;5:162–72.
Hermannsdörfer J, Friedrich M, Miyajima N, Albuquerque RQ, Kümmel S, Kempe R. Ni/Pd@MIL-101: synergistic catalysis with cavity-conform Ni/Pd nanoparticles. Angew Chem Int Edit. 2012;51(46):11473–7.
Maury F. Trends in precursor selection for MOCVD. Chem Vap Depos. 1996;2(3):113–6.
Hierso JC, Feurer R, Kalck P. Platinum, palladium and rhodium complexes as volatile precursors for depositing materials. Coord Chem Rev. 1998;178:1811–34.
Hämäläinen J, Ritala M, Leskelä M. Atomic layer deposition of noble metals and their oxides. Chem Mater. 2013;26(1):786–801.
Gozum JE, Pollina DM, Jensen JA, Girolami GS. “Tailored” organometallics as precursors for the chemical vapor deposition of high-purity palladium and platinum thin films. J Am Chem Soc. 1988;110(8):2688–9.
Zharkova GI, Stabnikov PA, Sysoev SA, Igumenov IK. Volatility and crystal lattice energy of palladium(II) chelates. J Struct Chem. 2005;46(2):320–7.
Zharkova GI, Sysoev SV, Stabnikov PA, Logvinenko VA, Igumenov IK. Vapor pressure and crystal lattice energy of volatile palladium(II) β-iminoketonates. J Therm Anal Calorim. 2011;103:381–5.
Basato M, Faggin E, Tubaro C, Veronese AC. Volatile square planar β-imino carbonyl enolato complexes of Pd(II) and Ni(II) as potential MOCVD precursors. Polyhedron. 2009;28(7):1229–34.
Brückmann L, Tyrra W, Stucky S, Mathur S. Novel air-stable and volatile bis(pyridylalkenolato) palladium(II) and-platinum(II) derivatives. Inorg Chem. 2011;51(1):536–42.
Assim K, Melzer M, Korb M, Rüffer T, Jakob A, Noll J, Georgi C, Schulz SE, Lang H. Bis (β-diketonato)-and allyl-(β-diketonato)-palladium(II) complexes: synthesis, characterization and MOCVD application. RSC Adv. 2016;6(104):102557–69.
Krisyuk VV, Sysoev SV, Turgambaeva AE, Nazarova AA, Koretskaya TP, Igumenov IK, Morozova NB. Thermal behavior of methoxy-substituted Pd and Cu β-diketonates and their heterobimetallic complex. J Therm Anal Calorim. 2017;130(2):1105–10.
Cherkasov SA, Vikulova ES, Nikolaea NS, Smolentsev AI, Morozova NB. Crystal structure and thermal properties of N,N’-(2,2-dimethylpropylene)-bis(acetylacetoniminato)palladium(II). J Struct Chem. 2017;58(7):1453–6.
McCarthy PJ, Hovey RJ, Ueno K, Martell AE. Inner complex chelates. I. Analogs of bisacetylacetoneethylenediimine and its metal chelates. J Am Chem Soc. 1955;77(22):5820–4.
Krisyuk VV, Tkachev SV, Baidina IA, Korolkov IV, Turgambaeva AE, Igumenov IK. Volatile Pd–Pb and Cu–Pb heterometallic complexes: structure, properties, and trans-to-cis isomerization under cocrystallization of Pd and Cu β-diketonates with Pb hexafluoroacetylacetonate. J Coord Chem. 2015;68(11):1890–902.
Bruker, APEX2 (Version 1.08), SAINT (Version 7.03), SADABS (Version 2.11) and SHELXTL (Version 6.12). Bruker AXS Inc., Madison, WI, USA; 2004.
Crystal Impact, DIAMOND (Version 3.2k), Crystal Impact GbR, Bonn, Germany; 2014.
Kubaschewski O, Evans EL. Metallurgical thermochemistry. London: Pergamon Press; 1951.
Volkova TV, Blokhina SV, Ryzhakov AM, Sharapova AV, Ol’khovich MV, Perlovich GL. Vapor pressure and sublimation thermodynamics of aminobenzoic acid, nicotinic acid, and related amido-derivatives. J Therm Anal Calorim. 2016;123(1):841–9.
Vikulova ES, Zherikova KV, Korolkov IV, Zelenina LN, Chusova TP, Sysoev SV, Alferova NI, Morozova NB, Igumenov IK. Thermal properties of mixed-ligand magnesium complexes with beta-diketonates and diamimes as potential MOCVD precursors. J Therm Anal Calorim. 2014;118(2):849–56.
Zherikova KV, Zelenina LN, Pishchur DP, Emel’yanenko VN, Shoifet E, Schick C, Verevkin SP, Gelfond VN, Morozova NB. Thermochemical study of rhodium(III) acetylacetonate. J Chem Thermodyn. 2016;102:442–50.
Dorovskikh SI, Kuratieva NV, Tkachev SV, Trubin SV, Stabnikov PA, Morozova NB. Copper(II) complexes with Schiff bases: structures and thermal behavior. J Struct Chem. 2014;55(6):1067–74.
Belcher R, Blessel K, Cardwell T, Pravica M, Stephen WI, Uden PC. Volatile transition metal complexes of bis-acetylacetone-ethylenedi-imine and its fluorinated analogues. J Inorg Nucl Chem. 1973;35(4):1127–44.
Dorovskikh SI, Filatov ES, Stabnikov PA, Morozova NB, Igumenov IK. N, N- and O, N-coordinated Co(II) β-diketonate derivatives: synthesis, structures, thermal properties and MOCVD application. Phys Proc. 2013;46:193–9.
Arockiasamy S, Premkumar PA, Sreedharan OM, Mallika C, Raghunathan VS, Nagaraja KS. TG/DTA-based techniques for the determination of equilibrium vapour pressures of N,N′-propylenebis(2,4-pentanedion-iminoato)nickel(II) for CVD applications. J Mater Sci. 2006;41:3383–90.
Premkumar PA, Pankajavalli R, Sreedharan OM, Raghunathan VS, Nagaraja KS, Mallika C. Determination of vapour pressure and standard enthalpies of sublimation and vapourisation of N,N′-ethylenebis(2,4-pentanedion-iminoato)nickel(II) by a TG-based transpiration method. Mater Lett. 2004;58:2256–60.
Zharkova GI, Sysoev SV, Turgambaeva AE, Igumenov IK. Thermal behavior of a series of monomeric Ni(II) complexes with β-iminoketones. Thermochim Acta. 2013;560:7–11.
Acknowledgements
The authors thank Federal Agency for Scientific Organizations for funding.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Vikulova, E.S., Cherkasov, S.A., Nikolaeva, N.S. et al. Thermal behavior of volatile palladium(II) complexes with tetradentate Schiff bases containing propylene-diimine bridge. J Therm Anal Calorim 135, 2573–2582 (2019). https://doi.org/10.1007/s10973-018-7371-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7371-z