
Abstract
Aluminum silicate nanofibers (ASNFs) have attracted significant attention due to their excellent stability and high-temperature properties. Al2O3-SiO2 aerogel (ASAs) has high porosity and low thermal conductivity, but its mechanical properties need to be improved. Reinforcing aerogels with fibers can lead to remarkable enhancements in their properties. Reducing the fiber diameter has proven to be an effective means of improving the structural integrity and mechanical stability of aerogel composites. In the context of this study, we prepared aluminum silicate nanofibers (ASNFs) with a diameter of 170 nm through the utilization of electrostatic spinning. These ASNFs were then successfully integrated with aluminum silicate aerogels (ASAs) to create a novel composite material known as aluminum silicate nanofiber-reinforced Al2O3-SiO2 aerogel (AS/ASNFAs). Its microstructure, mechanical properties and heat insulation properties have been researched. The results show that the compressive strength of AS/ASNFAs (0.44 MPa) is significantly higher than that of Al2O3-SiO2 aerogel (0.16 MPa). Meanwhile, the AS/ASNFAs has high specific surface area (600 m2/g), low density (0.15 g/cm3), and low thermal conductivity (0.026 W/(m·K)). This work provides a useful solution to improve the comprehensive properties of Al2O3–SiO2 aerogel composites.
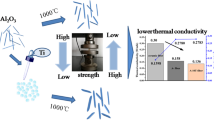
Graphical Abstract

Highlight
-
An aluminum silicate nanofiber-reinforced Al2O3-SiO2 aerogel (AS/ASNFAs) has been prepared by sol-gel method.
-
Addition of aluminum silicate nanofibers (ASNFs) improves mechanical properties of Al2O3-SiO2 aerogel (ASAs), and compressive strength of AS/ASNFAs-0.2 (0.44 MPa) is more than twice that of ASAs (0.16 MPa).
-
Low thermal conductivity (0.026 W/(m·K)) is achieved for AS/ASNFAs-0.1.
-
Due to the good dispersion of ASNFs in ASAs, the interaction of them not only improves mechanical properties of AS/ASNFAs but also increases thermal stability at high temperature.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Aerogel is a material with very high specific surface area, low density and porosity. The structure of the aerogel is similar to that of a sponge, with highly interconnected pores and a porous internal structure, which gives the aerogel extremely low density and ultra-high specific surface area [1,2,3,4], and it has been widely used in thermal insulation, adsorption, separation and photoelectric catalysis, etc [5]. Due to high heat resistance and high catalytic activity, Al2O3 aerogels have been considered as promising thermal insulators and catalyst carriers at high temperatures. However, poor mechanical properties and low phase stability at high temperature limit its utilization in high-temperature thermal fields [6, 7]. SiO2 doping can effectively inhibit the sintering and phase change of alumina aerogel, thereby improving its thermal stability [8]. Al2O3-SiO2 aerogel (ASAs) has better heat resistance and high porosity, and it has been widely used in catalysis, thermal insulation, and so on. However, ASAs nanoparticles are usually connected by small contact surfaces between particles (“pearl necklace” microstructure) [9], leading to limited mechanical properties. Thus, it is crucial to improve the mechanical properties of ASAs.
To improve the mechanical properties of ASAs, enhancing the strength of aerogel skeletons [10,11,12] and compounding with reinforcement materials [13, 14] are considered effective methods. The aerogel skeleton can be enhanced by surface regrowth or surface modification, but surface engineering requires significant aging time and multiple solution replacements. So far, inorganic fibers (such as SiO2, SiC, Al2O3 and ZrO2), organic polymer materials [15] (polyvinyl alcohol and polyethylene glycol), carbon nanotubes and graphene are widely used as reinforcing materials for aerogel. However, graphene, carbon nanotubes, polyvinyl alcohol (PVA) and polyethylene glycol (PEG) are easily oxidized at high temperatures, causing irretrievable damage to the aerogel structure. Inorganic fibers have become a good choice for their high strength and good resistance to oxidation [16]. However, there is a huge size difference between the fiber and aerogel (aerogel has a nano-scale skeleton structure and the fiber diameter is micron), thus aerogel is mostly divided into small pieces adhering to the fiber. Continuous nano-scale three-dimensional network structure of aerogel is changed by addition of fiber, so the advantages of aerogel such as high porosity and high specific surface area may be affected. Therefore, How to retain nano-scale skeleton structure of aerogel after addition of fibers is significative for fiber reinforced ASAs [17].
Nanofibers have excellent dielectric properties [18, 19], chemical stability [20], low thermal conductivity [21, 22] and excellent high-temperature properties [23, 24]. It has been shown that reducing the fiber diameter can improve the structural integrity and mechanical stability of aerogel composites [25,26,27]. Among the many nanofibers, aluminum silicate nanofibers (ASNFs) are ultra-light, high-temperature resistant, have excellent mechanical properties and possess better dispersibility in solution. Besides, the diameter of ASNFs is about 160 nm. If ASNFs are added into ASAs, the size difference between fibers (micrometer diameter) and aerogels (nanoscale skeletal structures) is low, good mechanical properties and heat insulation properties may be achieved. In this work, ASNFs are introduced into ASAs to prepare aluminum silicate nanofibers/Al2O3-SiO2 aerogel (AS/ASFAs), the effect of introducing ASNFs on the skeletal structure of ASAs is carefully researched. The effects of ASNFs on the microstructure, mechanical properties and thermal insulation properties of ASAs are also investigated.
2 Experimental
2.1 Materials
Aluminum isopropoxide (AIP), Tetraethyl orthosilicate (TEOS), N, N-dimethylformamide (DMF), Polyvinylpyrrolidone (PVP, MW = 1,300,000) were purchased from Shanghai Macklin, Aluminum nitrate (Al(NO)3·9H2O), Aluminum chloride (AlCl3·6H2O), 1, 2-epoxypropane (PO) were purchased from Shanghai Aladdin, Ethanol (EtOH) was purchased from Sinopharm, Hydrochloric acid (HCl, 36.0–38.0%) was purchased from Xilong Science Co. All of the chemicals were used as-received without further purification.
2.2 Preparation of aluminum silicate nanofibers (ASNFs)
AIP, Al(NO)3·9H2O, and TEOS were used as the aluminum and silicon sources. A certain amount of Al(NO)3·9H2O was dissolved in a mixed solution of EtOH and H2O, then AIP and TEOS were added under stirring conditions, stirred at room temperature for 24 h, followed by hydrolysis at reflux for 5 h at 85 °C. The molar ratio of AIP: Al(NO)3·9H2O: TEOS: EtOH: H2O was 2:1:1:25:25. PVP was dissolved in DMF with the mass fraction of 14 wt%. The alumina-silica sol and PVP/DMF solution were mixed in equal mass and stirred for 3 h at room temperature to obtain clear and transparent spinning precursors.
The nanofibers were prepared by electrostatic spinning with a 20-gauge stainless steel needle, a voltage of 21–23 kV, a collection distance of 18–20 cm, an injection rate of 0.8–1 ml/h, a humidity of 45–50%, and a temperature of 22–25 °C. The PVP/Aluminum Silicate Nanofiber films (PVP/ASNFs) were collected and dried overnight at 60 °C, followed by calcination at 800 °C for 2 h at a heating rate of 5 °C/min to obtain Aluminum Silicate nanofibers (ASNFs).
2.3 Preparation of aluminum silicate nanofiber/Al2O3-SiO2 aerogels (AS/ASNFAs)
Figure 1 shows the preparation process of AS/ASNFAs: TEOS and AlCl3·6H2O were used as the silicon and aluminum sources, respectively. The silica sol was synthesized by pre-hydrolyzing TEOS at 25 °C with a mass ratio of TEOS, H2O, EtOH and HCl of 20.8:17.5:1.8:0.13. The aluminum sol was obtained by pre-hydrolyzing AlCl3⋅6H2O at 25 °C with a mass ratio of AlCl3⋅6H2O, EtOH and H2O of 6:21:9. 1 g silicon sol was added to 10.864 g aluminum sol dropwise and kept stirring for 1 h to obtain the homogeneous aluminum-silicon sol, in which the molar ratio of Al to Si is 3:1, and a certain mass of crushed ASNFs was added and kept stirring for 1 h, and 2.5 ml 1,2-epoxypropane (PO) was added into the mixed solutions dropwise and the gel was usually completed within 3 h to obtain the AS/ASNFs gel. The gel was aged at 25 °C for 72 h in EtOH, with EtOH changes every 24 h. Finally, AS/ASNFAs were obtained by supercritical drying using CO2 as the drying agent. The samples were denoted AS/ASNFAs-X, where X represented the mass of ASNFs.

Scheme of AS/ASNFAs preparation
2.4 Characterizations
The bulk density of AS/ASNFAs-X composites is obtained by the ratio of mass to the volume, where the volume is gained by measuring the diameter and height of the samples with vernier caliper. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) was used to morphologies and elemental analysis of the samples (ZEISS, Gemini 300, Germany). Fourier transform infrared spectroscopy (ALPHAII, Germany) was performed to analyze surface compositions of the prepared samples. A surface area and pore size analyzer (JW-BK122W, China) was used to record N2 adsorption-desorption isotherms. Surface areas, pore volumes, and pore sizes were determined by the Brunauer-Emmett-Teller and Barrett-Joyner-Halenda methods. X-ray diffraction (TD-3500, China) was performed to determine the crystalline phases of the studied samples, while their thermal properties were investigated by differential scanning calorim-etry (DSC) and thermogravimetry (TG) in the temperature range from 50 to 1200 °C at a heating rate of 10 °C /min in flowing air using a thermal analyzer (NetzschSta449F5, Germany). Hot disk thermal analyzer (XIATECH, TC3200, China) was used to measure the room-temperature thermal conductivities of the samples. Electronic universal testing machine (SUNS, UTM4304X, China) was used to measure the compressive strengths of the samples.
3 Results and discussion
3.1 Morphology and elemental composition of PVP/ASNFs, ASNFs
The morphology of ASNFs and PVP/aluminum silicate nanofiber (PVP/ASNFs) are shown in Fig. 2. Both PVP/ASNFs and ASNFs have normal cylindrical morphology and continuous smooth surfaces (Fig. 2a–d). The fabricated PVP/ASNFs film is stretched and knotted without being destroyed, indicating its excellent flexibility and stretchability (Fig. 2f). Figure 2c, d shows the morphology of ASNFs obtained by calcination of PVP/ASNFs. Due to the removal of solvents and organics during calcination [28], the average diameter of calcined nanofibers decreases from 273 nm to about 170 nm. Because prepared nanofibers have continuous homogeneity, they need to be broken before use. The crushing results are shown in Fig. 2e, where the nanofibers are broken into short-cut fibers of uneven length. EDS results suggest the uniform distribution of silica and alumina in ASNFs (Fig. 2g–i).

Morphological and elemental characterizations of PVP/ASNFs and MNFs: a, b, f The appearance and microscopic structures of PVP/ASNFs; c, d SEM of ASNFs; g–j EDS of ASNFs
The morphology of ASAs and AS/ASNFAs are shown in Fig. 3. As shown in Fig. 3a, b, ASAs has a slender backbone, and it presents as a continuous three-dimensional network structure. The ASNFs are embedded in the ASAs and encapsulated by the ASAs backbone, which is analogous to the reinforced concrete structure in buildings. Besides, the addition of ASNFs does not disrupt the matrix structure of ASAs (Fig. 3c–e). Due to capillary forces during the drying process and condensation reactions of hydroxyl groups (-OH) within the ASAs, significant localized internal stresses are generated, rendering the ASAs susceptible to defects and cracks (Fig. 7c). However, the ASNFs are uniformly dispersed within the ASAs and establish effective contact points, mitigating the substantial internal stresses arising within the ASAs. This results in a smooth and gentle passage through the drying stage for AS/ASNFAs, thereby preserving their continuous and intact structure (Figs. 3f and 7c). This indicates that the inclusion of ASNFs helps prevent cracking due to shrinkage during drying, ultimately benefiting the mechanical properties of the composite.. The physical properties of ASAs and AS/ASNFAs are listed in Table 1. The density of ASAs, AS/ASNFAs-0.1 and AS/ASNFAs-0.2 are 0.1583 g/cm3, 0.1549 g/cm3 and 0.1612 g/cm3. This suggests the addition of ASNFs has little effect on density of ASAs, and AS/ASNFAs have ultra-light weight. Besides, the EDS results show a uniform distribution of aluminum and silicon in AS/ASNFAs (Fig. 3g–i).

Morphological and elemental characterizations of ASAs and AS/ASNFAs: a, b ASAs; c–e AS/ASNFAs; f Optical images of AS/ASNFAs; g–j EDS of AS/ASNFAs
3.2 Pore structure and chemical construction of ASAs and AS/ASNFAs
The pore size distribution, nitrogen adsorption and desorption isotherms of ASAs and AS/ASNFAs samples are shown in Fig. 4a–c. The relevant datas are listed in Table 1. There are apparent hysteresis loops of ASAs and AS/ASNFAs at relative pressures of 0.8–1.0 in Fig. 4a–c, and a short platform appears at relative pressures near 1.0. The nitrogen adsorption and desorption isotherms of ASAs and AS/ASNFAs are consistent with IV curves in the IUPAC classification. The hysteresis loops of the AS/ASNFAs exhibit H3-type characteristics, indicating that both ASAs and AS/ASNFAs are mesoporous materials with a lamellar structure [9, 17, 29, 30]. When the content of ASNFs increases from 0 to 0.2, the average pore size and pore volume increase first and then decrease (Fig. 4a–c, Table 1). The specific surface area of the AS/ASNFAs decreases from 685.972 m2/g to 562.232 m2/g, which is caused by the space occupied by ASNFs. This is consistent with the SEM of AS/ASNFAs (Fig. 3c–e). Table 1 also shows that all AS/ASNFAs samples exhibit low thermal conductivity at room temperature (0.026 W/(m·K)).

a–c Nitrogen adsorption-desorption isotherms and pore size distribution of ASAs, AS/ASNFAs-0.1 and AS/ASNFAs-0.2; d FT-IR curves of ASAs and AS/ASNFAs between 4000 and 400 cm−1
The FT-IR spectra of ASAs and AS/ASNFAs samples between 4000 and 400 cm−1 at room temperature are shown in Fig. 4d. The absorption peaks at 3400–3500 cm−1 and 1634 cm−1 represent the stretching and bending vibrations of the -OH groups from water and EtOH [9, 30,31,32]. The peaks at 2900–3000 cm−1 are due to the stretching and bending vibrations of -CH groups [29, 33]. The peaks at 530 and 880 cm−1 represent the vibrations of the Al-O bonds of the boehmite [10, 34, 35]. The peaks at 1389 and 1340 cm−1 represent the vibrations of AlO-H bonds of the boehmite [36]. The peaks at 1042 cm−1 correspond to Al-O-Si stretching vibrations caused by the formation of -Al-O-Si- groups in the gel structure [31, 36]. The FT-IR spectra of the AS/ASNFAs are similar to the ASAs, suggesting ASNFs does not affect the structure of ASAs.
3.3 Thermal performance of ASNFs and AS/ASNFAs
3.3.1 Morphology of ASNFs and AS/ASNFAs during heat treatment
Thermal stability is significant for aerogel. The evolution of ASNFs after heat treatment is shown in Fig. 5a–d. There are white spots in ASNFs after heat treatment at 1000 °C, which is caused by the generation of the mullite phase. The diameter of ASNFs becomes significantly smaller after heat treatment at 1200 °C and the mullite grains appear. The microstructure of ASAs after heat treatment at 800, 1000, 1200, and 1300 °C are shown in Fig. 5e–h. After heat treatment at 800 °C, the specific surface area of ASAs decreases. Although ASAs have a complete skeleton structure, there are some cracking and fragmentation (Fig. 7c), which is detrimental to the application of aerogel. After heat treatment at 1000 °C, the fragile skeletal structure start to sinter. When heat treatment temperature increase to 1300 °C, severe sintering occurs. The microstructure of AS/ASNFAs after heat treatment at 800, 1000, 1200, and 1300 °C are shown in Fig. 5i–l. After heat treatment at 800 °C, ASNFs are submerged in the ASAs skeletal structure. AS/ASNFAs retain intact and continuous skeletal structure, and fragmentation is quite avoided (Fig. 7c). After heat treatment at 1000 °C, AS/ASNFAs has slender skeletal structure. This indicates that the addition of ASNFAs slow down mullitization of alumina and silica. When heat treatment temperature increases to 1200 °C, ASNFs act as scaffold-like structure to alleviate the sintering. After treatment at 1300 °C, the AS/ASNFAs sinters severely. However, there are no mullite spots and grains in the ASNFs, indicating ASAs simultaneously slowes down the evolution of mullitization in the ASNFs.

SEM images of ASNFs, ASAs and AS/ASNFAs after heat treatment. a, b SEM images of ASNFs after heat treatment at 1000 °Cfor 2 h; c, d SEM images of ASNFs after high treatment at 1200 °Cfor 2 h; e–h SEM images of ASAs after high treatment at 800, 1000, 1200, and 1300 °C for 2 h; i–l SEM images of AS/ASNFAs after high treatment at 800, 1000, 1200, and 1300 °C for 2 h
3.3.2 Pore structure of ASNFs and AS/ASNFAs during heat treatment
The specific surface area of ASAs and AS/ASNFAs after heat treatment are shown in Fig. 6a. The specific surface area of AS/ASNFAs decreases with the increment of heat treatment temperature. The AS/ASNFAs still have high specific surface area after the heat treatment at 800 °C, indicating that the heat treatment at 800 °C does not cause damage to the skeleton structure of AS/ASNFAs. The specific surface area of AS/ASNFAs decreases after heat treatment at 1000 °C, which is caused by the generation of the mullite phase [9, 30]. The specific surface area of AS/ASNFAs is only about 40 m2/g after heat treatment at 1200 °C due to sintering. The adsorption and desorption isotherm curves of AS/ASNFAs samples after different temperature treatments are shown in Fig. 6b–d. The AS/ASNFAs after different temperature treatments exhibit similar isotherm curves (type IV isotherms with H3-type hysteresis loops), indicating that AS/ASNFAs are mainly mesoporous materials with layered pores [29, 37].

a Specific surface area of ASAs and AS/ASNFAs samples after high-temperature treatment; b–d Nitrogen adsorption-desorption isotherms and pore size distribution of ASAs and AS/ASNFAs samples after heat treatment at 800, 1000, and 1200 °Cfor 2 h, respectively
The images of AS/ASNFAs samples after heat treatment at 600 and 800 °C are shown in Fig. 7. The linear shrinkage of the samples after heat treatment was 21.54% and 25.98%. The main reason is the degradation of the organic components and the condensation reaction of the free hydroxyl group (-OH). AS/ASNFAs are intact after heat treatment at 800 °C without cracking and fragmentation phenomena. This may be because the structure of AS/ASNFAs is similar to the concrete, ASNFs act as a steel structure to resist stretching and good stability is achieved. After calcination with butane flame for 1 minute, surface temperature of AS/ASNFAs increases. Calcination of organic matter and the condensation reaction of hydroxyl (Si-OH, Al-OH) lead to cracks in surface of AS/ASNFAs. It is noteworthy that the cracks only appear on the front side surface of AS/ASNFAs, while the back side of AS/ASNFAs remains continuous and dense. This suggests that the composites have certain temperature resistance. In a word, the AS/ASNFAs exhibit complete morphology in the different temperature environments, suggesting good stability of aerogels.

a1–a3 The optical images of the AS/ASNFAs samples after heat treatment at 600 and 800 °Cfor 2 h; b1, b2 The optical images of AS/ASNFAs after calcination in butane flame for 1 min; c1, c2 The optical images of ASAs and AS/ASNFAs and the optical images of ASAs and AS/ASNFAs samples after heat treatment at 800 °C
3.3.3 Thermal behavior and crystalline structure of ASNFs and AS/ASNFAs during heat treatment
TG-DSC has been used to study the thermal behavior of ASNFs and AS/ASNFAs. As shown in Fig. 8a, PVP/ASNFs have two mass loss stages. Before 200 °C (stage I), the volatilization of adsorbed water and the release of organic substances are the main factors of mass loss. From 200 to 650 °C (stage II), the thermal oxidative decomposition of organic components of PVP. After 650 °C, the mass is relatively constant. The DSC curve shows there is a clear exothermic peak around 991 °C, which is caused by mullitization (3Al2O3 + 2SiO2 → 3Al2O3·2SiO2) [28]. This is consistent with formation of mullite grains on surface of ASNFs (Fig. 5b). The TG-DSC curve of ASAs is shown in Fig. 8b. The weight loss and endothermic peak below 400 °C are primarily caused by the desorption of physically adsorbed water and decomposition of the residual organics. The exothermic peak at 310 °C belongs to decomposition of residual alkoxy (-OC2H5) and 1,2-epoxypropane from ASAs [9, 30]. The broad exothermic peak appearing at 550 °C is caused by the structural transition from pseudo-boehmite toγ-Al2O3 (2AlO(OH)→γ-Al2O3 + H2O) [30], and the exothermic peak appearing at 1009 °C is the result of mullitization [34, 38]. The TG-DSC curve of AS/ASNFAs-0.1 shows two exothermic peaks near 1000 °C, which is corresponding to mullitization of ASNFs and ASAs. This indicates that the addition of ASNFs does not affect the ASAs structure.

a TG-DSC curves of PVP/ASNFs; b, c TG-DSC curves of ASAs and AS/ASNFAs samples; d XRD patterns of ASNFs after heat treatment at 800, 1000, and 1200 °C for 2 h, respectively; e, f XRD patterns of ASAs and AS/ASNFAs after heat treatment at 800, 1000, 1200, and 1300 °Cfor 2 h, respectively
XRD was used to study the structural changes of ASNFs and AS/ASNFAs during heat treatment, and the results are shown in Fig. 8d–f. As shown in Fig. 8d, the XRD pattern of ASNF shows diffraction peaks after heat treatment at 1200 °C, corresponding to the mullite phase (PDF#79-1453) [34]. This agrees with the appearance of grains in SEM (Fig. 5c, d). The XRD pattern of ASAs sample shows mullite phase [17] (PDF#79-1276) diffraction peaks after heat treatment at 1300 °C (Fig. 8e). Notably, the AS/ASNFAs transitions to the mullite phase (PDF#79-1276) after heat treatment at 1200 °C, which is produced by mullitization of ASNFs. Nanofibers sinter in the ASAs skeleton structure also confirms this (Fig. 5k, l).
3.3.4 Mechanical property of AS/ASNFAs
The compressive stress-strain curves of the AS/ASNFAs are shown in Fig. 9. The compressive stress of ASAs is 0.16 MPa at 7 % strain. The compressive stress of AS/ASNFAs-0.1 reaches 0.22 MPa. This suggests the addition of ASNFs improve the mechanical property of ASAs. In particular, the AS/MNFAs-0.1 (density of 0.15 g/cm3) with a weight of 0.51 g could withstand up to ~1000 times their weight without breaking. As the content of ASNFs increases, the compressive stress of AS/ASNFAs-0.2 reaches 0.44 MPa. This is attributed to the load generated by the aerogel adhering to the more fiber surface [9]. The compressive stress of AS/ASNFAs-0.1 after high treatment at 800 °Cis 1.22 MPa, indicating that the AS/ASNFAs-0.1 have higher compressive resistance after heat treatment. Overall, the addition of ASNFs improved the compressive strength of the pure aerogels, and the AS/ASNFAs samples exhibited higher compressive strength after heat treatment.

Compressive stress-strain curves of the ASAs and the AS/ASNFAs
4 Conclusion
In this work, aluminum silicate nanofibers (ASNFs) were fabricated by the electrostatic spinning method, and it was used to fabricate aluminum silicate nanofiber/Al2O3-SiO2 aerogels (AS/ASNFAs). The addition of ASNFs increases compressive strength of the ASAs, and compressive strength of AS/ASNFAs-0.2 (0.44 MPa) is significantly higher than that of ASAs (0.16 MPa). Meanwhile, the AS/ASNFAs-0.1 has high specific surface area (600 m2/g), low density (0.15 g/cm3), and low thermal conductivity (0.026 W/(m·K)). Due to the good dispersion of ASNFs in Al2O3-SiO2 aerogel, the interaction between ASNFs and Al2O3-SiO2 aerogel not only improves mechanical properties of composites but also increases thermal stability at high temperature. This work is a useful exploration to design aerogel/nanofiber composites with good mechanical properties and low thermal conductivity, and it can be potentially applied in high temperature thermal insulations and catalysis.
References
Wu SH, Mou CY, Lin HP (2013) Synthesis of mesoporous silica nanoparticles. Chem Soc Rev 42(9):3862–3875. https://doi.org/10.1039/c3cs35405a
Almeida CMR, Ghica ME, Duraes L (2020) An overview on alumina-silica-based aerogels. Adv Colloid Interface Sci 282:102189. https://doi.org/10.1016/j.cis.2020.102189
Mazrouei-Sebdani Z, Begum H, Schoenwald S, Horoshenkov KV, Malfait WJ (2021) A review on silica aerogel-based materials for acoustic applications. J Non Crystalline Solids 562. https://doi.org/10.1016/j.jnoncrysol.2021.120770
Linhares T, Pessoa de Amorim MT, Durães L (2019) Silica aerogel composites with embedded fibres: a review on their preparation, properties and applications. J Mater Chem A 7(40):22768–22802. https://doi.org/10.1039/c9ta04811a
Xia C, Hao M, Liu W, Zhang X, Miao Y, Ma C, Gao F (2023) Synthesis of Al2O3-SiO2 aerogel from water glass with high thermal stability and low thermal conductivity. J Sol-Gel Sci Technol 106(2):561–571. https://doi.org/10.1007/s10971-023-06085-y
Zu G, Shen J, Zou L, Wang W, Lian Y, Zhang Z, Du A (2013) Nanoengineering super heat-resistant, strong alumina aerogels. Chem Mater 25(23):4757–4764. https://doi.org/10.1021/cm402900y
Shi Z, Gao H, Wang X, Li C, Wang W, Hong Z, Zhi M (2018) One-step synthesis of monolithic micro-nano yttria stabilized ZrO2-Al2O3 composite aerogel. Microporous Mesoporous Mater 259:26–32. https://doi.org/10.1016/j.micromeso.2017.09.025
Slosarczyk A (2017) Recent advances in research on the synthetic fiber based silica aerogel nanocomposites. Nanomaterials 7 (2). https://doi.org/10.3390/nano7020044
Yu H, Tong Z, Yue S, Li X, Su D, Ji H (2021) Effect of SiO2 deposition on thermal stability of Al2O3-SiO2 aerogel. J Eur Ceram Soc 41(1):580–589. https://doi.org/10.1016/j.jeurceramsoc.2020.09.015
Zu G, Shen J, Wang W, Zou L, Lian Y, Zhang Z, Liu B, Zhang F (2014) Robust, highly thermally stable, core–shell nanostructured metal oxide aerogels as high-temperature thermal superinsulators, adsorbents, and catalysts. Chem Mater 26(19):5761–5772. https://doi.org/10.1021/cm502886t
Wu L, Huang Y, Wang Z, Liu L, Xu H (2010) Fabrication of hydrophobic alumina aerogel monoliths by surface modification and ambient pressure drying. Appl Surf Sci 256(20):5973–5977. https://doi.org/10.1016/j.apsusc.2010.03.104
Liu B, Gao M, Liu X, Zhao X, Zhang J, Yi X (2019) Thermally stable nanoporous ZrO2/SiO2 hybrid aerogels for thermal insulation. ACS Appl Nano Mater 2(11):7299–7310. https://doi.org/10.1021/acsanm.9b01791
Barrios E, Fox D, Li Sip YY, Catarata R, Calderon JE, Azim N, Afrin S, Zhang Z, Zhai L (2019) Nanomaterials in advanced, high-performance aerogel composites: a review. Polymers (Basel) 11 (4). https://doi.org/10.3390/polym11040726
Yang Z, Zhu D, Li H (2020) A chitosan-assisted co-assembly synthetic route to low-shrinkage Al2O3–SiO2 aerogel via ambient pressure drying. Microporous and Mesoporous Materials 293. https://doi.org/10.1016/j.micromeso.2019.109781
Yang K, Venkataraman M, Karpiskova J, Suzuki Y, Ullah S, Kim I-S, Militky J, Wang Y, Yang T, Wiener J, Zhu G, Yao J (2021) Structural analysis of embedding polyethylene glycol in silica aerogel. Microporous Mesoporous Mater 310. https://doi.org/10.1016/j.micromeso.2020.110636
Yi Z, Zhang X, Yan L, Huyan X, Zhang T, Liu S, Guo A, Liu J, Hou F (2022) Super-insulated, flexible, and high resilient mullite fiber reinforced silica aerogel composites by interfacial modification with nanoscale mullite whisker. Composites Part B Eng 230. https://doi.org/10.1016/j.compositesb.2021.109549
Liu L, Wang X, Zhang Z, Shi Y, Zhao Y, Shen S, Yao X, Shen J (2022) A facile method for fabricating a monolithic mullite fiber-reinforced alumina aerogel with excellent mechanical and thermal properties. Gels 8 (6). https://doi.org/10.3390/gels8060380
Awang N, Nasir AM, Yajid MAM, Jaafar J (2021) A review on advancement and future perspective of 3D hierarchical porous aerogels based on electrospun polymer nanofibers for electrochemical energy storage application. J Environ Chem Eng 9 (4). https://doi.org/10.1016/j.jece.2021.105437
Chhetri K, Subedi S, Muthurasu A, Ko TH, Dahal B, Kim HY (2022) A review on nanofiber reinforced aerogels for energy storage and conversion applications. J Energy Storage 46. https://doi.org/10.1016/j.est.2021.103927
Cheng X, Liu YT, Si Y, Yu J, Ding B (2022) Direct synthesis of highly stretchable ceramic nanofibrous aerogels via 3D reaction electrospinning. Nat Commun 13(1):2637. https://doi.org/10.1038/s41467-022-30435-z
Guo J, Fu S, Deng Y, Xu X, Laima S, Liu D, Zhang P, Zhou J, Zhao H, Yu H, Dang S, Zhang J, Zhao Y, Li H, Duan X (2022) Hypocrystalline ceramic aerogels for thermal insulation at extreme conditions. Nature 606(7916):909. https://doi.org/10.1038/s41586-022-04784-0
Xian L, Zhang Y, Wu Y, Zhang X, Dong X, Liu J, Guo A (2020) Microstructural evolution of mullite nanofibrous aerogels with different ice crystal growth inhibitors. Ceram Int 46(2):1869–1875. https://doi.org/10.1016/j.ceramint.2019.09.163
Zhang B, Liu Y, Wu Q, Zhou M, Su D, Ji H, Li X (2022) Super-insulating, ultralight and high-strength mullite-based nanofiber composite aerogels. J Eur Ceram Soc 42(13):5995–6004. https://doi.org/10.1016/j.jeurceramsoc.2022.06.061
Liu R, Dong X, Xie S, Jia T, Xue Y, Liu J, Jing W, Guo A (2019) Ultralight, thermal insulating, and high-temperature-resistant mullite-based nanofibrous aerogels. Chem Eng J 360:464–472. https://doi.org/10.1016/j.cej.2018.12.018
Zhang R, An Z, Zhao Y, Zhang L, Zhou P (2020) Nanofibers reinforced silica aerogel composites having flexibility and ultra‐low thermal conductivity. Int J Appl Ceram Technol 17(3):1531–1539. https://doi.org/10.1111/ijac.13457
Wu H, Chen Y, Chen Q, Ding Y, Zhou X, Gao H (2013) Synthesis of flexible aerogel composites reinforced with electrospun nanofibers and microparticles for thermal insulation. J Nanomater 2013:1–8. https://doi.org/10.1155/2013/375093
Zimmermann MVG, Zattera AJ (2021) Silica aerogel reinforced with cellulose nanofibers. J Porous Mater 28(5):1325–1333. https://doi.org/10.1007/s10934-021-01080-6
Wang J, Liu W, Song X, Ma Y, Huang Y (2018) Effects of added polyvinyl pyrrolidone on morphology and microstructure of multiple-phase mullite nanofibers. Ceram Int 44(13):15418–15427. https://doi.org/10.1016/j.ceramint.2018.05.195
Jiang D, Qin J, Zhou X, Li Q, Yi D, Wang B (2022) Improvement of thermal insulation and compressive performance of Al2O3–SiO2 aerogel by doping carbon nanotubes. Ceram Int 48(11):16290–16299. https://doi.org/10.1016/j.ceramint.2022.02.178
Peng F, Jiang Y, Feng J, Li L, Cai H, Feng J (2020) A facile method to fabricate monolithic alumina–silica aerogels with high surface areas and good mechanical properties. J Eur Ceram Soc 40(6):2480–2488. https://doi.org/10.1016/j.jeurceramsoc.2020.01.058
Yu H, Jiang Y, Lu Y, Li X, Zhao H, Ji Y, Wang M (2019) Quartz fiber reinforced Al2O3-SiO2 aerogel composite with highly thermal stability by ambient pressure drying. J Non Crystalline Solids 505:79–86. https://doi.org/10.1016/j.jnoncrysol.2018.10.039
Yang Z, Li H, Niu G, Wang J, Zhu D (2021) Poly(vinylalcohol)/chitosan-based high-strength, fire-retardant and smoke-suppressant composite aerogels incorporating aluminum species via freeze drying. Composites Part B Eng 219. https://doi.org/10.1016/j.compositesb.2021.108919
Zhang X, Zhang R, Jin S, Hu Z, Liu Y, Jin M (2018) Synthesis of alumina aerogels from AlCl3·6H2O with an aid of acetoacetic-grafted polyvinyl alcohol. J Sol Gel Sci Technol 87(2):486–495. https://doi.org/10.1007/s10971-018-4679-x
Wu X, Shao G, Shen X, Cui S, Wang L (2016) Novel Al2O3–SiO2composite aerogels with high specific surface area at elevated temperatures with different alumina/silica molar ratios prepared by a non-alkoxide sol–gel method. RSC Adv 6(7):5611–5620. https://doi.org/10.1039/c5ra19764c
Wen S, Ren H, Zhu J, Bi Y, Zhang L (2018) Fabrication of Al2O3 aerogel-SiO2 fiber composite with enhanced thermal insulation and high heat resistance. J Porous Mater 26(4):1027–1034. https://doi.org/10.1007/s10934-018-0700-6
Cai H, Jiang Y, Chen Q, Zhang S, Li L, Feng J, Feng J (2020) Sintering behavior of SiO2 aerogel composites reinforced by mullite fibers via in-situ rapid heating TEM observations. J Eur Ceram Soc 40(1):127–135. https://doi.org/10.1016/j.jeurceramsoc.2019.09.014
Peng F, Jiang Y, Liu F, Feng J, Feng J, Li L (2022) Hydrothermal assisted synthesis of heat resistant, well-crystallized aerogels constructed by boehmite nano rods. Ceram Int 48(11):16232–16240. https://doi.org/10.1016/j.ceramint.2022.02.171
Wu X, Shao G, Cui S, Wang L, Shen X (2016) Synthesis of a novel Al2O3–SiO2 composite aerogel with high specific surface area at elevated temperatures using inexpensive inorganic salt of aluminum. Ceram Int 42(1):874–882. https://doi.org/10.1016/j.ceramint.2015.09.012
Acknowledgements
Authors thank to Research Project of Shanxi Scholarship Council of China (Grant No. 2022-042), Key R & D program of Shanxi Province (Grant No. 202102030201006), Shanxi Province Porous Ceramic Material Technology Innovation Center (Grant No. 202104010911002) and the Program of applied basic research program of shanxi province (Grant No. 202103021223055)
Author contributions
MH plays a role in methodology, software, investigation, writing-original draft, validation, formal analysis; HC and CX takes charge of formal analysis; TS takes charge of visualization; CM plays a guiding role in conceptualization, review editing and supervision; YM takes charge of data curation, supervision.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hao, M., Chen, H., Xia, C. et al. Al2O3-SiO2 aerogel reinforced with aluminum silicate nanofibers: a strategy to preserve the properties of Al2O3-SiO2 aerogel. J Sol-Gel Sci Technol 109, 523–533 (2024). https://doi.org/10.1007/s10971-023-06286-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-023-06286-5