
Abstract
For mechanical properties of titanium, oxygen content is important. In re-certifying O mass fraction in the Ti certified reference material ERM-EB090b, instrumental photon activation analysis (IPAA) was used as an alternative to the inert gas fusion technique. IPAA utilizes 15O measurable only via the nonspecific annihilation gamma line 511 keV. Interferences from other radionuclides can be suppressed by optimizing photon energy and irradiation-decay-counting times, and for most interfering radionuclides corrected via their specific gamma lines. The present re-evaluated IPAA results for O content 3.56 ± 0.59 g/kg matches closely the assigned certified value 3.57 ± 0.19 g/kg.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Titanium is a metal with outstanding mechanical and chemical properties such as high specific strength, corrosion resistance, and biocompatibility, which make it a suitable construction material for, e.g., medical devices or bone implants. For mechanical and chemical properties of Ti, besides trace metal impurities, contents of light elements C, H, N, and O are important [1,2,3,4]. In 2018, European Commission Joint Research Centre, Directorate F–Health, Consumers and Reference Materials produced the certified reference materials (CRM) ERM-EB090a and ERM-EB090b (Titanium), and reported on certified values for trace metal contents and information values for C, H, N, and O [5]. In 2019–2020, an interlaboratory comparison was organized to get quality data for certifying also the mass fractions of C, H, N, and O. For C assay, combustion combined with IR spectrometry was used exclusively. For N, H and O assays, the inert gas fusion technique (IGF) was used followed by thermal conductivity detection (N) and infrared detection (H and O) by most participants. As alternatives, two nuclear analytical methods were employed–prompt gamma activation analysis for H assay, and instrumental photon activation analysis (IPAA) for O assay [6]. The IPAA result for O content in ERM-EB090b 4.07 ± 0.61 g/kg was within uncertainty limits in agreement with the assigned certified value 3.57 ± 0.19 g/kg. Yet, the somewhat higher IPAA result, by about 14%, led us to speculations about uncorrected unseen interferences in IPAA, or about possible loss of O due to sorption onto flux impurities in IGF mentioned in the ASTM Standard Test Method E 1409 [7]. The latter explanation seems quite improbable, regarding generally good consistency (i.e., low scatter around the mean value) of the independent IGF data from various participating laboratories [6].
In preparation of a presentation for the RANC 2023 conference, the IPAA data and their evaluation were thoroughly revised. A probable cause of the overvaluation has been tracked down in improper correction of a time-dependent interference contribution from Mo. The present paper describes in detail all experimental and corrected evaluation procedures applied in the IPAA assay of O in the Ti CRM ERM-EB090b, utilizing irradiation with < 23 MeV bremsstrahlung photons produced by the MT 25 microtron.
Theory
In principle, photon activation analysis is capable of determining C, N and O via their short-lived photoactivation products 11C, 13N and 15O, pure positron emitters measurable only via the annihilation gamma line 511 keV. The nonspecific annihilation radiation is interfered by number of other positron emitting radionuclides. Photoactivation assay of those light elements, mostly in pure metals or steel, has been employed since pioneering works on photon activation analysis in 1950s (see their reviews in [8, 9] and references therein). In those works, the assay of the analyte nuclides usually required their radiochemical (i.e., post-activation) separation from the sample matrix.
In the instrumental, (quasi) nondestructive mode (i.e., IPAA), the interferences could be suppressed by optimizing photon energy and irradiation-decay-counting times thanks to large differences in the threshold energies of photonuclear reactions or in half-lives of their products. For the interfering radionuclides which are not pure positron emitters and have also specific gamma lines, contributions to the 511 keV line can be evaluated and subtracted. In favorable or optimized cases, deconvolution of the multicomponent decay function is possible. We successfully applied such approaches previously in the assay of N and F [9,10,11], and adopted the simple equations presented there similarly for 15O assay in Ti; see below Eqs. (1–3).
A proportion of 15O in the total 511 keV peak area counted in a sample can be obtained by subtracting all significant interference contributions using the equation
An individual interference contribution from an interferent i (see Table 1) is evaluated by multiplying its specific peak area counted in the sample with a correction factor CFi, which is calculated as
The correction factors are obtained by dividing the 511 keV peak area by the specific peak area counted in an irradiated pure standard of the given interferent, using the same detector with the same counting geometry as for samples and O calibrator.
In the case of corrections for Ti as the main interferent, because of quite a low intensity of the 45Ti main specific line 719.6 keV, counting the auxiliary line 159.4 keV of 47Sc produced by photoactivation of 48Ti (see Table 1) is more effective. As the two different products of Ti photoactivation have quite different half-lives (see Table 1), the correction factors for 45Ti derived via 47Sc are time-dependent according to the equation
Here CF(t1) and CF(t2) are the correction factor values in decay times t1 and t2 (t2 > t1), respectively, and λ47Sc and λ45Ti are decay constants of 47Sc and 45Ti, respectively.
Throughout the study in evaluation of parameters derived from counting, a great care had to be taken for proper corrections for decay after irradiation and during counting using the decay and counting factors D and C, respectively, defined in a standard way (see [8] and [13], e.g.) as
Here λ is a decay constant of a respective radionuclide, td is a decay time between the end of irradiation and start of counting, and tc is a counting time. In the correction, a counted peak area is divided by D and/or C.
Experimental
Preparation of samples and standards
All titanium pellets were dipped in concentrated HF:HNO3 1:4 for 5–10 s, then rinsed 3 times with deionized water, then with aceton, and dried. From all pellets, 3 sample aliquots of a comparable mass (ca. 1.4 g each) were prepared using 5 or 6 pellets packed in an aluminum foil, in “5 on a playing cube” or “pentagon flower” layouts, respectively. Additional measurement and calculation of an average sample thickness was done to make necessary corrections for varying sample thickness.
The final calibration standard was done from cellulose (Fluka 22,182). For the cellulose calibration standard, a perfect stoichiometry according to the formula C6H10O5 has been assumed. Three standard aliquots were prepared as pressed pellets (d = 16 mm) with various mass (128–333 mg O) and thickness (0.9–2.3 mm).
The correction standards to derive correction factors CFi for Fe and Mo (and other elements for which interference turned out to be negligible) were prepared as flat samples from pure metal powders (283 and 108 mg Fe and Mo, respectively), packed in a thin PE foil. Correction of the Ti interference was done directly from sample counting.
Irradiation and counting
The MT 25 microtron, a high frequency cyclic accelerator of electrons (see [14] for technical parameters) was employed as a source of high energy photons for IPAA irradiations. The high energy photon radiation is produced by braking the accelerated electrons in a water cooled tungsten converter. Unfortunately, due to technical problems of the magnetron with expiring lifetime, the mean electron current of only 6 μA was achieved with the electron beam energy set at 23 MeV. Also, a pneumatic tube delivery system for rapid sample transport between the irradiation position and detector installed at MT 25 was out of operation, and the irradiations had to be carried out in the offline mode requiring to turn off MT 25, open the armoured shielding doors and transport irradiated samples to the detector manually.
Samples, as well as calibration and correction standards, were irradiated individually, positioned directly on the front of the beam stopper (3.5 cm thick aluminum plate placed after the tungsten converter). Irradiation of each sample or standard took ~ 3 min to reach a photon dose identical for each sample, which was controlled by fine adjustment of irradiation time until total charge of accelerated electrons reached 4.3 mC. The corresponding mean photon fluence rate was ~ 4 × 1011 cm−2 s−1 for photons between 16 and 23 MeV, i.e., over the threshold energy of 15O production and below the maximum photon energy equal to the accelerated electron beam energy.
An irradiated sample was removed from packing foil and repacked into a clean polyethylene bag. The necessary operations including sample transport, repacking and placing to detector delayed the minimum achievable decay time before counting to ~ 150 s. For gamma counting, an HPGe detector Canberra-Packard GR3020 (CP, 30%, reverse, Be window, FWHM 2.3 at 1332 keV), connected to a Lynx® Digital Signal Analyzer was used. A “flat” sample (all pellets in one layer) was counted 1 cm from the detector top. The correction standards were irradiated and counted under similar conditions (but for decay–counting times) as samples and standards. The following (initial) decay/counting time regimes were applied: CRM ERM-EB090b (Ti) sample (150 s/10 × 60 s and 60 min/1 × 60 s), calibrator (150 s/10 × 60 s and 60 min/1 × 60 s), Mo (150 s/4 × 60 s), Fe (400 s/3 × 60 s). Counted areas of the 511 keV peak of both samples and standards were sufficiently high for a low counting uncertainty (< 1%).
Results and discussion
Pilot tests
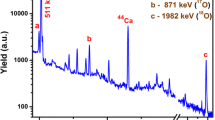
Preliminary calculations and pilot tests indicated that trace amounts of C and N in Ti are below their IPAA detection limits in CRM ERM-EB090b, and only O determination via 15O should be feasible. Inspection of gamma spectrum from a pilot photoactivation of CRM ERM-EB090b indicated the presence of possibly interfering nuclides (besides 45Ti as the major interferent) 53Fe, 63Zn 89mZr, 91mMo, and 95Ru. Except for Zn, contents of their target elements in CRM ERM-EB090b are reported as certified values in the certification report [5]. An optimum irradiation time of 3 min based on activity build up relative to the saturation activity has been chosen. It has been found that in the optimized irradiation-decay-counting time regime only 54Fe and 92Mo interference contributions can be evaluated, and the other ones are probably negligible.
Calibration
Cellulose used as an O calibrator contains C interfering via its photoactivation product 11C. However, 11C half-life is ten times longer than that of 15O (cf. in Table 1), thus its known contribution can easily be corrected. No other impurities have been identified. After 3-min irradiation, counting started with a 3-min delay as a sequence of ten 1-min counting runs. An additional 1-min counting was added after 60-min decay (15O completely decayed), which was used to calculate retrospective activities of 11C in individual counting runs to be subtracted from the total 511 keV peak area. A semi-logarithmic time plot of the obtained pure 15O peak area showed perfect linearity (R2 = 1.00) with a derived half-life (2.04 min) very close to the theoretical value. The derived calibration constant (peak area per μg) is dependent on the calibrator pellet thickness. Thus, the calibration was done on three pellets with different thickness (0.9, 1.8, and 2.3 mm), and a 3-point thickness dependence (linear, R2 = 1.00) was evaluated for matching the calibration constant to sample thickness. Also, a lower gamma self-attenuation in cellulose compared to Ti (by 2% for 2 mm sample thickness) has been accounted for, based on calculation from published mass attenuation coefficients for Ti, C, H, and O at 662 keV (137Cs), for cellulose applying a mixture rule [15,16,17].
Correction factors (CF)
Determination of CF for 45Ti as the major interferent was done directly during irradiation and counting of the three available sample aliquots (see the Experimental). It used an auxiliary gamma line 159 keV of 47Sc produced by photoactivation of 48Ti; see above Eq. (3) with the related discussion. Counting started ca. 150 s after the 3-min irradiation as a sequence of ten 1-min counting runs. An additional 1-min counting was added after ca. 60 min when 15O and short-lived interfering nuclides sufficiently decayed. The CF values were calculated as a ratio of 511 keV peak areas to those at 159 keV, corrected by common decay and counting factors according to Eqs. (4) and (5), respectively. The CF values are higher at few early counting runs due to presence of 15O and other short-lived interfering nuclides. Mean CF values were calculated from the last (additional) counting runs by averaging for 3 aliquots to give a value of 7.4. Because CF is calculated from activities of two radionuclides with quite different half-lives, it has to be decay-corrected using Eq. (3) before using for calculation of pure 45Ti contributions to 511 keV line. In fact, the 45Ti contributions calculated via CF corresponded very well (within 2%) with the 511 keV peak areas (corrected by decay and counting factors) obtained for the additional counting run, indicating that virtually only 45Ti was present after 60-min decay.
The CF values for Fe and Mo were calculated according to Eq. (2) as the ratios of peak areas at 511 keV to those at 378 keV (53Fe) and 653 keV (91mMo) measured in respective correction standards. The correction standards could also be used as calibrators for determining Fe and Mo contents in CRM ERM-EB090b. Such attempt has been done and the results (Fe 1.59 g/kg, Mo 0.533 g/kg) agreed quite well with the certified values (Fe 1.82 g/kg, Mo 0.484 g/kg [5]). In the case of CF for Fe, a mean 6.93 ± 0.02 was obtained from three subsequent countings. In the case of Mo, the CF value was strongly time-dependent, probably due to production of two radionuclides, 91mMo and 91Mo, being moreover in genetic relation. No specific gamma lines of 91Mo were detected. The time dependence of the four CF values determined via the 653 keV line of 91mMo was fitted with a simple exponential growth function (R2 = 1.00). The CF value 1.87 obtained by extrapolation to zero time is not far from a ratio of 511 to 653 keV photon intensities (76/53 = 1.43; [12]), regarding the counting efficiency of an HPGe detector decreasing with energy. In the former evaluation of the IPAA data for the certification [6], the CF value extrapolated to zero time was applied by mistake instead of a value fitted to an appropriate decay time, leading to undervaluation of the Mo interference contribution. This error has been corrected in the present re-evaluation.
Determination of O content
The mass fraction of O in CRM ERM-EB090b was determined from the results of the irradiation and counting runs as described above in determination of CF for 45Ti. The measured 511 keV peak area is time dependent, following a multiple exponential decay function composed of several components decaying with different rates (45Ti, 15O, and all minor interfering nuclides). The large number of components makes it difficult to fit an exact function, thus for the decay correction during counting using Eq. (5), a “virtual” decay constant was evaluated from peak areas in each two subsequent counting runs. The 45Ti, 53Fe, and 91mMo–91mMo contributions were evaluated from specific peak areas (corrected for decay during counting) and CF values derived as described in the respective section above, and subtracted from the decay corrected 511 keV peak area according to Eq. (1). The resulting pure 15O peak area was corrected for total decay from the end of irradiation using Eq. (4), and together with the average sample thickness, and thickness and self-attenuation corrected calibration constant (see the sections on sample preparation and calibration) used for calculation of O content in sample. From the 6 experimental results (2 counting runs on 3 sample aliquots), the mean value ± 2σ of 3.56 ± 0.59 g/kg has been obtained for the O mass fraction in CRM ERM-EB090b (2σ represents the population standard deviation expanded to 95% confidence interval). In Table 2, a concise analytical scheme including the calibration, interference correction, and sample analysis stages has been summarized.
IPAA performance within the interlaboratory comparison
Comparison of the IPAA result for the O mass fraction in CRM ERM-EB090b achieved in the present study with those obtained within the certification interlaboratory comparisons [5, 6] including the previous questionable IPAA result with the erroneous Mo interference correction, is illustrated by Fig. 1. The present IPAA result 3.56 ± 0.59 g/kg matches closely the assigned certified value 3.57 ± 0.19 g/kg [6]. On the other hand, it is obvious that the uncertainty associated with the IPAA value (2σ ~ 17 rel.%) is higher than the uncertainty of the certified value (~ 5 rel.%) and those generally declared for IGF results (~ 1–6 rel.%, except for ~ 20 rel.% declared by the lab L4 [6]).

Comparison of the IPAA results for the O mass fraction in CRM ERM-EB090b with those obtained by the inert gas fusion technique with infrared detection (IGF-IR) within the interlaboratory comparisons [5, 6]. The current and previous IPAA results are depicted by a square and circle, respectively. The pre-certification IGF-IR values [5] are marked with asterisks in the labcodes and depicted by grey triangles. The assigned certified value and uncertainty (3.57 ± 0.19 g/kg [6]) are depicted by dashed and dotted lines, respectively
Despite of counting the 511 keV line with good statistics (area uncertainty < 1 rel.%), uncertainties in determining numerous, mostly time-dependent interference corrections were generally higher, and have been propagated by their combination. Also, the fact that the sample matrix is composed of Ti as the major interferent has not been favourable. A 15O fraction of the 511 keV peak area was only about 9–10% and 6–7% in the first and second counting runs, respectively. The Fe and Mo interference contributions were each about 2% of the 511 keV peak area in both counting runs, and their slower decrease with decay time increased their proportion in later counting runs. Performance of the IPAA procedure thus must have been significantly deteriorated by the excessively long decay time between the end of irradiation and start of counting, which could be reduced if the pneumatic transport system was available.
Conclusions
Potential of instrumental photon activation analysis (IPAA) in determining trace amounts of oxygen in metallic titanium has been demonstrated within the interlaboratory comparison organized to complete certification of the Ti certified reference material ERM-EB090b for the elements C, H, N, and O. An optimized IPAA procedure, based on < 23 MeV bremsstrashlung irradiation and counting the nonspecific annihilation gamma line 511 keV of the photoactivation product 15O, included 3-min irradiation, 3-min decay, and a sequence of 1-min counting runs. Evaluation of the O mass fraction consisted in decay curve analysis and deconvolution of the 511 keV line, with subtraction of interferences from photoactivation of Ti as a major constituent, and Mo and Fe impurities, each evaluated via respective correction factors. The correction factors were obtained as ratios of the 511 keV line to a specific gamma line of a respective interfering radionuclide; for Ti directly from irradiation and counting of an analysed sample as an internal standard, for Mo and Fe from separate irradiation and counting of external standards. Attention has to be paid to proper correction of all time-dependent quantities, namely the Ti and Mo correction factors. To maximize the 15O and minimize the interference proportions in the analytical gamma peak, minimizing decay time between the end of irradiation and start of counting is crucial.
Data availability
The data presented in the study are openly available in references stated or on request from the corresponding author.
References
Kariya S, Fukuo M, Umeda J, Kondoh K (2019) Quantitative analysis on light elements solution strengthening in pure titanium sintered materials by Labusch model using experimental data. Mater Trans 60:263–268. https://doi.org/10.2320/matertrans.Y-M2018849
He WJ, Zhang SH, Song HW, Cheng M (2009) Hydrogen-induced hardening and softening of a β-titanium alloy. Scr Mater 61:16–19. https://doi.org/10.1016/j.scriptamat.2009.02.030
Luo SD, Song T, Lu SL, Liu B, Tian J, Qian M (2020) High oxygen-content titanium and titanium alloys made from powder. J Alloys Compd 836:155526. https://doi.org/10.1016/j.jallcom.2020.155526
Donato TAG, de Almeida LH, Arana-Chavez VE, Grandini CR (2014) In vitro cytotoxicity of a Ti-35Nb-7Zr-5Ta alloy doped with different oxygen contents. Materials 7:2183–2193. https://doi.org/10.3390/ma7032183
Bacquart T, Linsinger TPJ (2018) The certification of the mass fraction of elements in titanium: ERM®-EB090a and ERM®-EB090b. Eur Comm Joint Res Centre Directorate F-Health Consum Ref Mater Geel Belgium. https://doi.org/10.2760/180660
Linsinger TPJ (2020) The certification of the mass fractions of C, H, N and O in the certified reference materials ERM®-EB090a and ERM®-EB090b. Eur Comm Joint Res Centre Directorate F-Health Consum Ref Mater Geel Belgium. https://doi.org/10.2760/287817
ASTM E 1409 (1997) Standard test method for determination of oxygen in titanium and titanium alloys by the inert gas fusion technique. American Society for Testing and Materials, West Conshohocken
Segebade C, Berger A (2008) Photon Activation Analysis. In: Meyers RA (ed) Encyclopedia of Analytical Chemistry. Wiley, Hoboken. https://doi.org/10.1002/9780470027318.a6211.pub2
Mádlíková M, Krausová I, Mizera J, Táborský J, Faměra O, Chvátil D (2018) Nitrogen assay in winter wheat by short-time instrumental photon activation analysis and its comparison with the Kjeldahl method. J Radioanal Nucl Chem 317:479–486. https://doi.org/10.1007/s10967-018-5881-6
Krausová I, Mizera J, Řanda Z, Chvátil D, Krist P (2015) Nondestructive assay of fluorine in geological and other materials by instrumental photon activation analysis with a microtron. Nucl Instrum Methods Phys Res Sect B 342:82–86. https://doi.org/10.1016/j.nimb.2014.09.023
Krausová I, Mizera J, Dostálek P, Řanda Z (2018) Non-destructive determination of nitrogen in malting barleys by instrumental photon activation analysis and its comparison with the Dumas method. J Inst Brew 124:4–8. https://doi.org/10.1002/jib.477
Řanda Z, Kreisinger F (1983) Tables of nuclear constants for gamma-activation analysis. J Radioanal Chem 77:279–495. https://doi.org/10.1007/BF02522230
Krausová I, Mizera J, Řanda Z, Chvátil D, Krist P (2021) Instrumental photon activation analysis with short-time irradiation for geochemical research. Minerals 11:617. https://doi.org/10.3390/min11060617
Krist P, Horák Z, Mizera J, Chvátil D, Vognar M, Řanda Z (2015) Innovation at the MT-25 microtron aimed at applications in photon activation analysis. J Radioanal Nucl Chem 304:183–188. https://doi.org/10.1007/s10967-014-3578-z
Goswami B, Chaudhuri N (1973) Measurements of gamma-ray attenuation coefficients. Phys Rev A 7:1912–1916. https://doi.org/10.1103/PhysRevA.7.1912
Makarewicz M (2005) Estimation of the uncertainty components associated with the measurement of radionuclides in air filters using γ-ray spectrometry. Accred Qual Assur 10:269–276. https://doi.org/10.1007/s00769-005-0931-5
Eke C (2023) Gamma-ray attenuation characteristics of various chemical fertilizers. Instrum Exp Tech 66:111–118. https://doi.org/10.1134/S0020441223010098
Acknowledgements
The authors thank Thomas P. J. Linsinger of the EC JRC for the invitation to participate in the interlaboratory comparison.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mizera, J., Krausová, I., Chvátil, D. et al. Oxygen determination in the Ti certified reference material ERM-EB090b by instrumental photon activation analysis. J Radioanal Nucl Chem 333, 3481–3486 (2024). https://doi.org/10.1007/s10967-023-09260-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-023-09260-1