
Abstract
Just 1 year after the Budapest Research Reactor went critical on 29 March 1959, the Neutron Activation Analysis laboratory started its operation. During the past 55 years this laboratory contributed to many important innovations in NAA and gamma spectroscopy, including the k 0-standardization, irradiation-site characterization, detector efficiency calibration and gamma spectroscopy. From 2012, the laboratory is being revitalized and instrumentally modernized. This paper gives a comprehensive overview of the present technical capabilities, the ongoing upgrade program and the prospects for future utilization. The analysis protocols are discussed in details to give guidance to our user community.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neutron activation analysis (NAA) is a method for quantitative composition analysis of chemical elements based on the conversion of stable nuclei in the sample to radioactive nuclei by nuclear reactions, followed by the quantitation of the reaction products via their gamma radiations. The k 0-standardized neutron activation analysis (k 0-NAA) [1], combined with high-resolution and high-throughput gamma-ray spectrometry, offers mostly non-destructive, multi-elemental analysis for many branches of science and technology, such as environment [2, 3], geochemistry [4], microelectronics, power plant technology [5, 6], biology and health science [7, 8], forensics applications, archaeometry and material science [9]. In these studies we benefit from its inherently favorable characteristics, e.g. negligible matrix effect, excellent selectivity and high sensitivity. NAA is a precise and accurate panoramic analysis technique that has recently been recognized by metrologists as a primary ratio method [10]. Even small amount (few tens of milligrams) of samples (mostly solid) can be measured, in which about 35–75 elements in less than 0.01 μg quantities can be determined. Relative standardization method remains also an option for high-precision analysis of selected components in known matrices.
The Budapest Research Reactor (BRR) is one of the leading research infrastructures in Hungary and even in Central-Europe, presently managed by the Centre for Energy Research, Hungarian Academy of Sciences (formerly KFKI, and KFKI-AEKI). The neutron activation analysis laboratory started its operation here already in 1960. The BRR is a tank type VVR (light water-cooled, light water-moderated reactor) Soviet-designed and built reactor. The reactor had initially a thermal power of 2 MW, fueled with EK-10 fuel assemblies with 10 % enrichment. Between 1967 and 1986, it ran at 5 MW with VVR-SzM type fuel (36 % enrichment) and got an additional beryllium reflector, resulting in a maximum in-core thermal equivalent neutron flux of 4.5 × 1013 n cm−2 s−1. This was the era when most of the k 0-standardisation [11, 12] measurements took place, in collaboration between A. Simonits and the group at University of Ghent led by F. De Corte. From 1986 to 1992 a full-scale upgrade was carried out, supported by the International Atomic Energy Agency (IAEA) and the European Union (EU). The new, 10 MW-reactor was commissioned on 12 December 1992 and expected to operate annually about 3000 h until 2023. Since 2009, thanks to a core-conversion program, 19.8 % enriched VVR-M2 type fuel is used. In 1997, Budapest Neutron Centre (BNC) was founded to co-ordinate and to promote research activities and scientific utilization of the reactor. BNC, and more specifically the NAA laboratory, participates in several transnational access programs like NMI3, C-ERIC and IPERION CH.
The Budapest neutron activation analysis laboratory was recently unified with the prompt gamma activation analysis, radiography and Mössbauer groups—formed the Nuclear Analysis and Radiography Department. This reorganization opened the way to the synergetic utilization of PGAA and NAA. The complementary nature of the techniques in terms of amenable elements and detection limits make them an ideal couple, especially in the analysis of metals and geological samples.
Experimental
Irradiation sites
One of the 40 vertical irradiation channels in the research reactor is equipped with a fast rabbit system operated directly from the laboratory. This is made of 50 m long flexible polypropylene tube with photo detectors along the capsule path to have feedback about the capsule’s position and speed. The last five meter of the pipe, i.e. the in-core part is made of aluminum. The rabbit system ensures that the irradiated sample goes from the reactor zone to the gamma spectrometer in the shortest possible time. The fluctuation of thermal neutron flux is <5 % along the longitudinal axis of the irradiation zone and <0.5 % inside the internal polyethylene capsules used for irradiating the individual samples (10 mm long, 8 mm diameter). At present configuration, irradiations from 10 s to 5 min can be carried out using a polyimide (DuPont™ Vespel SP-1) carrier capsule. The block-scheme of the fast rabbit system is shown in Fig. 1.

The block scheme of the fast rabbit system. 1 Sender/receiver station (IFÁ), 2 Router, 3 Delay stacker, 4 Air tank, 5 Reactor vessel, 6 Drop-out station
Powder samples for short irradiations are packed in a Whatman 41 filter paper, and pressed in pellet form of 6 mm diameter and 3 mm height, using a hydraulic press. Sometimes dissolution and homogenization is necessary. For liquids well-defined volumes of the solution (10–100 μl) are deposited on a carrier material (e.g., high purity filter paper or Al foil) surface using a micropipette, dried with an infrared lamp and finally the pastilles are made. A state-of-the-art Mettler-Toledo XPE 26 microbalance was installed to the accurate weighing (<0.7 μg reproducibility) of the tiny samples (typically 5–10 mg) as well as the accompanying flux monitors. Pellets then placed in cleaned polyethylene capsules for irradiation. After irradiation the sample pellets are repacked into inactive polyethylene, and measured for 10 min after 2–10 min decay time, and optionally, at a second time for 20–30 min. The quantitative analyses of the elements Al, Ca, Cl, Cu, Dy, In, K, Mg, Mn, Na, Sm, Ti, V are possible on a routine basis from the short-term irradiation.
Long-term irradiation (typically 12–24 h) is done separately, in the No. 17 rotating channel located in the Be reflector. Samples, in dried and powdered form (50–200 mg), are sealed in high-purity quartz ampoules (6 cm long, 6 mm ∅, Suprasil AN, Heraeus), whose inner and outer surfaces are etched in a mixture of hydrogen fluoride, acetic-, nitric acid and H2O2, washed with double distilled water and dried. A set of 6–8 quartz ampoules are put together using Al wires, as well as monitor foils of Au, Zr or Ni. Samples are irradiated in hermetically closed Al carriers. After irradiation and a typical decay time of 3–4 days, the quartz vials are unpacked and surface-cleaned. Alternatively, samples can be packed directly in high-purity aluminum too. Each sample is counted for two or three times. First measurement takes place at about half an hour after the sample arrives at the counting laboratory, when the elements As, Br, Ca, K, La, Na, Sb, U can be determined. The second, optional measurement takes place 5–10 days later (for 1–2 h), if the detection limits are needed to be improved, i.e. during this period the 24Na, 42K isotopes decay out. After a cooling period of 15–30 days (when the 82Br, 140La isotope decayed out), the samples can be counted for a third time for 2–6 h at appropriate distance, when typically Ba, Ce, Co, Cr, Cs, Eu, Fe, Rb, Sb, Sc, Ta, Tb, Yb, Zn, Zr elements are analyzed.
Neutron flux parameters are measured for each irradiation with the “Bare Triple-Monitor” method [13, 14] using Zr, Al-0.1 % Au and Fe foils thickness (0.025–0.1 mm) and the 94Zr, 96Zr and 197Au isotopes The thermal neutron flux of the fast-rabbit irradiation channel is 5.3 × 1013 n cm−2 s−1, it is well thermalized (f ≈ 37) and the α value is slightly positive under the common irradiation conditions (0.003–0.012). The fast neutron flux was measured to be 5.9 × 1012 n cm−2 s−1. The vertical, rotating channel for long irradiations has somewhat lower thermal neutron flux, 2.0 × 1013 n cm−2 s−1, a slightly higher f (≈47) and identical α. The fast neutron flux here is 1.1 × 1012 n cm−2 s−1. It is common that the nominal flux changes by 5–10 % due to rearrangement and burn-out of the fuel elements, so in-situ flux determination is needed for every irradiation. Special neutron fields are also applicable upon request, such as epicadmium [15] and epiboron [16] irradiations. The induced activation of the samples is predicted on a routine basis using the NAAPro [17] software.
Gamma-ray spectrometry
The gamma-rays emitted from the samples are counted in low-level counting chambers to reduce the external background. In our NAA laboratory there are low-level counting chambers of about 80 × 80 × 80 cm sizes, built from pre-World War II steel [18] with 10 cm wall thickness covered by 2 mm Cu-layer from inside. The samples can be placed at different distances from the detector face (0–30 cm in 5 cm increments), using a set of polycarbonate spacers.
By setting up a new counting station (see Fig. 2), now three upright-standing high-purity Ge detectors, each with a 30-l liquid nitrogen Dewar and connected to a digital gamma spectrometer are at our disposal to collect the gamma spectra. There are two p type detectors, ORTEC PopTop 55195-P HPGe (“D5”, 55 % rel. efficiency, energy resolution 0.95 keV @ 59.5 keV, 1.75 keV @ 1332 keV, RC feedback preamp), and Canberra GC3618 HPGe (“D4”, 36% rel. efficiency, energy resolution 0.80 keV @ 59.5 keV, 1.80 keV @ 1332 keV, TRP preamp) and an n type Canberra GC1318 HPGe (“D3”, 13 % rel. efficiency, energy resolution 0.95 keV @ 59.5 keV, 1.80 keV @ 1332 keV, RC feedback preamp) in routine use. Each detector was X-rayed to obtain the accurate dimensions and placements of the crystal within the detector cap for solid angle calculations.

The new counting station “D5” with the ORTEC DSPEC 502 dual input digital spectrometer
So far the detector signals were fed into spectrometers built from analog NIM modules (Canberra 2026 amplifier (4 μs shaping), 8715 8 k/800 ns ADC, ACCUSPEC/B MCA (2 × 8 k) card, Loss-Free Correction module (LFC), Genie 2000). These pieces of electronics were aging and therefore no longer reliable, they were hard to maintain, became incompatible with the recent computers and had performance bottlenecks in terms of count rate tolerance and energy resolution, therefore it was decided to adopt the recent digital signal processing technology. After careful testing of the available models from different manufacturers (see the “Results and discussion” section), the most suitable option was purchased, installed and fine-tuned. The 55 and 36 % detectors are now connected to a dual-input ORTEC DSPEC 502 spectrometer, whereas the 13 % HPGe is coupled to an ORTEC DSPEC+ unit. All spectrometers are operated by the ORTEC Maestro 7 software. The spectra, now with 2 × 16 k channel instead of the 2 × 8 k, are recorded with the zero-dead time (ZDT) option enabled. The corrected and uncorrected halves of the spectra are stored in a single SPC file. The SPC files can be converted to MCA, the primary file format of Hypermet PC, by a custom utility for further analysis or used in its native format.
Data analysis
The gamma peak fitting was done for 15 years using the DOS-based Hypermet-PC (ver. 5.) [19], which was developed jointly by the Budapest PGAA and NAA groups in the late nineties. Recently, its windows-based successor, HyperLab 2013.1 [20] was introduced in our analytical procedure. The batch fitting feature of Hyperlab makes the spectrum evaluation much more productive.
The spectrometers were accurately recalibrated at multiple distances for the energy range from 50 to 3300 keV, using 137Cs, 60Co, 152Eu, 226Ra, 133Ba, 207Bi, 241Am certified sources and several reactor-irradiated radionuclides (e.g. 24Na, 134Cs, 110mAg; 51Cr, 95Zr, 198Au). The FWHM, efficiency and nonlinearity data were set up in Hyperlab database. A method based on Monte Carlo calculations with MCNP5 was worked out to determine the activities of bulky or irregular-shaped samples.
For quantitative analysis, a home-written software called RNAACNC [21] was used. This software has modules for detector efficiency, absolute- and specific-activity calculation, thermal and epithermal neutron flux, or flux ratio (f) determination, alpha computation, isotope identification based on a nuclear data library, and calculation of element concentrations. It had however its limitations (geometry, nuclear data) and it is no longer maintained, therefore the introduction of other software based on k 0-standardization, i.e. the well-established k 0-IAEA 7.16 [22] and KayZero for Windows 3.06 [23] programs, were considered.
Results and discussion
Comparison of the digital spectrometers
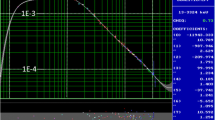
We compared the digital spectrometers Canberra Lynx and ORTEC DSPEC 502 with the old NIM-based electronics in terms of energy resolution, peak shape, count rate tolerance. We found that the Lynx did provide very similar resolution to our analog system, whereas with the DSPEC about 0.15-keV better value could be achieved at 1332 keV, when using the 50–3300 keV energy range (Fig. 3). This performance gain is attributed mostly to the filter function’s Tilt functionality [24] that compensates for the non-ideal behavior of the preamplifier signal. With Tilt set to zero, its resolution was quite similar to the other two systems. We found that setting Rise time to 8.4 μs and Flat top to 1.0 μs is a good compromise between count-rate tolerance and energy resolution for both detectors, with Tilt value of −0.046 (“D4”) and −0.125 (“D5”), respectively.

The energy resolution of the “D4” system with three different signal processing chains
It is interesting to note that the two inputs of our dual-channel DSPEC 502 unit shows remarkably different nonlinearity curves, but both could be handled adequately by Hyperlab.
We experimentally validated the trueness of the ZDT under our experimental conditions, using the two-source method [25], monitoring a 137Cs-peak at 661 keV. Our results confirmed the earlier literature conclusions [26] that the dead-time correction is accurate, equally for Canberra Lynx and Ortec DSPEC 502, up to about 85 % dead times (corresponding to about 800,000 cps) with both our detectors, equipped with RC Feedback (“D5”) and transistor reset (“D4”) preamplifiers. With the analog system, this count rate limitation was at about 50 % dead time. Under these extreme conditions, however, the distortion of the peak shape might limit the trueness and precision. The list mode feature of the ORTEC device seems to be well applicable to follow the time evolution of the peaks.
Comparison of analysis software
The two computer programs, KayZero for Windows and k 0-IAEA were used in parallel [27] to evaluate a set of mostly geological standard reference materials. We used z score statistics and the E n values defined in ISO/IEC 17025:2005 to assess the laboratory performance. It could be stated that with adequate care both programs can produce high-quality results (E n < 1) for most of the elements, but the uncertainty estimation of KayZero for Windows seems to be more consistent and there are less outliers (in contrast, Yb came out with E n numbers between 7 and 13 with k 0-IAEA). The results demonstrated in Fig. 4 are for the synthetic multi-element standard material, SMELS [28], that was prepared with the intention to have a reference material to validate the proper implementation of the k 0-NAA method in a laboratory. The data, measured with the two counting systems, D4 and D5, evaluated with both software, show clearly that our analytical procedure is in general fit for purpose. Due to better reliability and better-documented implementation of the original k 0-methodology, we will rely on KayZero for Windows in our final procedure.

The E n values for SMELS I, II and III using KayZero for Windows and k 0-IAEA. (Yb and Tm outliers with k 0-IAEA are shown truncated to fit into the graph)
Validation of the protocol with round-robin exercises
To validate our analysis procedure and assess the expertise of the staff we regularly participate in round-robin exercises. In the 2015 run of the WEPAL ISE intercomparison series we got a “consolidated good performance” classification. From the re-analysis of the 2013 sample Clay River basin (WEPAL ISE 870) [29] we concluded that the “new method” (new detector, digital signal processing, Hyperlab, KayZero for Windows, new analyst) is compatible with the “old method” (old detector, NIM electronics, Hypermet PC, RNAACNC, experienced analyst), but often provides lower uncertainties and better z scores. The detailed results are tabulated in Table 1.
As result of all the above efforts, our laboratory was also well-ranked in the 2015 Molybdenum intercomparison study organized by the TU Delft and the IAEA, where bulky pieces of metallic Mo (1 and 5 g pieces) and powdered MoO3 (m = 1 g) were irradiated and the thermal capture cross-section was to be determined. In order to arrive at the good value, the whole analysis procedure must be under control. One should well characterize the irradiation channel, measure the flux, handle the timing, calibrate the detector, correct for thermal and epithermal self-shielding, gamma-ray absorption, and propagate the uncertainty correctly. Our values for the three samples (0.129–0.131 barns ±5 %) were in excellent agreement with the reference literature data.
Future prospects
With increasing challenges from competing analytical techniques, such as ICP-OES, ICP-MS, as well as from laboratory- or synchrotron-based XRF, the demand for NAA started to decrease. In this evolving situation NAA has to find its new application niche, where the labor-intensive and more expensive analysis is acceptable and can still outperform other analytical techniques. An emphasis has to be put on its key properties, such as non-destructivity, representativeness, reliability and analysis of “exotic” elements in “exotic” matrices, where matrix-matched standards are too expensive or even unavailable. On the other hand, the applications shall focus on society-relevant topics, such as cultural heritage, environment, waste management, health industry.
Our vision about the sustained utilization of Budapest NAA and PGAA laboratories is the wise combination of NAA and PGAA techniques, in order to take advantage from the synergies. These are as follows:
-
When measuring the same sample with PGAA and NAA, one can detect nearly all elements from H to U in a totally unknown sample, from ppb levels to major components. The set of elements amenable to PGAA and NAA are often complementary (e.g. H, B, C, N, P, S, Si, Pb), if not, NAA still offers analysis possibility far below the detection limits of PGAA. For example, in geological samples, PGAA gives precise results for major elements (Si, Ti, Al, Fe, Mn, Mg, Ca, Na, K and H), for some trace elements (e.g. B and Cl), as well as for Sc, S, Cr, Co, Ni, Cd, Nd, Sm and Gd. NAA is sensitive for its rare earth elements, and for many major (Ti, Al, Fe, Mn, Mg, Ca, Na, K) and trace elements (e.g. Sc, V, Cr, Co, Ni, Cu, Zn, As, Se, Rb, Sr, Zr, As, Sb, Ce, Ba, Hf, Ta, W). In glass matrix, PGAA can well measure B, Na, Al, Si, Cl, K, Ca, Ti, Nd, Gd, Pb, whereas NAA is able to determine trace elements such as As, Ba, Br, Cd, Co, Cr, Fe, Ir, La, Mo, Sb, Se, Sn, Sr, Ta, Zn. In metals, e.g. bronzes, the dynamic range of the PGAA is often limited, so NAA is preferred for Fe, As, Sb, Zn quantification, but it cannot measure Pb. For most major elements our results obtained by the two techniques show good agreement [4]. On the other hand, PGAA seems to be more reliable to measure Al, Cl, Ca and Mn as major and minor components.
-
Furthermore, use can be made of our Olympus Delta Premium handheld XRF device for quick screening or for elements not well analyzed with either of the two activation techniques (e.g. low concentrations of Pb).
-
In NAA, one usually plans the irradiation conditions, calculates the sample weights, irradiation times based on a presumed composition. Unknown samples may still contain unexpected components that can be seriously activated during irradiation, reaching dose rates where manipulation of the sample is no longer permitted and it has to be discarded. To avoid such problems, one can rely on a PGAA result, where the presence of most problematic components can already be revealed.
-
Significant analytical bias can arise in NAA from invisible elements with high neutron-capture cross-section (e.g. boron) or high gamma-attenuation (e.g. Pb), due to the self-shielding or self-attenuation, respectively. PGAA, however, can very well detect B and Pb, and provide input to correct the NAA-measured elemental masses. This is especially useful in the analysis of glasses.
-
PGAA can provide more reliable results if NAA determination relies on a minor isotope, such as in case of sulfur or chlorine.
-
At least for geological samples, the set of elements amenable to short-term NAA and PGAA are very similar. A PGAA measurement at hand can make the short irradiation unnecessary, if the interesting components are above the detection limits of PGAA. This can spare manpower and analysis cost.
-
Technically, the analyses by PGAA and NAA are easiest to complete in parallel on two aliquots if possible. If only one sample is available, or it is particularly important to analyze the identical sample, than PGAA shall be done first to assess the major components, and after a few hours/days, when the activity from the in-beam irradiation has decayed out, the sample can be further analyzed for trace elements by NAA.
-
There seems to be an increasing need to determine accurately the nuclear parameters of some so-far unstudied nuclides (Gen IV reactor structural materials, actinides [30], etc.). This calls not just for a refinement, but for an extension of the NAA cross-section- and k 0-database.
-
In a guided beam of cold or thermal neutrons, especially if it is modulated with a beam chopper, or with a separate off-line counting [31], one can measure neutron capture cross sections or k 0-factors under a different condition than they were originally established. The harmonization of the nuclear datasets for PGAA and NAA can reveal inconsistencies and lead to a more correct analytical results from both techniques.
-
The combination of active and inactive tracing can provide significant contribution to control and develop industrial processes, as well as answer scientific questions.
-
The lab, and in general the whole reactor, will play an important role to sustain our nuclear culture, domestic and international educational and training activities, as Hungary committed itself for the nuclear energy production on a long term basis. For this purpose, new training program for MSc and PhD students was initiated.
Conclusions
After decades of success, followed by a generation change in staff and significant upgrade in the instrumentation, the NAA laboratory of the Budapest Neutron Centre has carried out a full review of its analysis protocol. The procedure was validated with the measurement of standard reference materials and round-robin exercises. These confirmed our competence to do reliable element analysis based on the k 0-methodology. After the upgrade program, thanks to the above described developments, and working in close collaboration with the PGAA instrument, the NAA laboratory became once again suitable to accept commercial requests, users from transnational user access programs, and serve the in-house applied research.
References
Simonits A, Decorte F, Hoste J (1975) Single-comparator methods in reactor neutron-activation analysis. J Radioanal Chem 24(1):31–46
Salma I, Zemplén-Papp E (1999) Instrumental neutron activation analysis for studying size-fractionated aerosols Nucl. Instrum Method A 435(3):462–474
Salma I, Maenhaut W, Zemplén-Papp E et al (2001) Comprehensive characterisation of atmospheric aerosols in Budapest, Hungary: physicochemical properties of inorganic species. Atmos Environ 35(25):4367–4378
Gméling K, Simonits A, Sziklai-László I et al (2014) Comparative PGAA and NAA results of geological samples and standards. J Radioanal Nucl Chem 300(2):507–516
Nagy P, Vajda N, Sziklai-László I et al (2014) Determination of Cs-135 in nuclear power plant wastes by ICP-MS and k 0-NAA. J Radioanal Nucl Chem 300(2):615–627
Sziklai-László I, Kocsis E, Nyeste J et al (1976) Determination of chloride and sodium concentrations in high-purity waters. Radiochem Radioanal Lett 25(2):129–136
Ördögh M (1978) Complex neutron-activation method for analysis of biological-materials. J Radioanal Chem 46(1):27–40
Szőke R, Alföldi B, Balásházy I et al (2007) Size distribution, pulmonary deposition and chemical composition of Hungarian biosoluble glass fibers. Inhal Toxicol 19(4):325–332
Rausch H, Braun T (1997) Multielemental characterization of several brands of fullerenes and fullerene precursors by instrumental neutron activation analysis. Anal Chem 69(13):2312–2316
Greenberg R, Bode P, De Nadai Fernandes E (2011) Neutron activation analysis: a primary method of measurement. Spectrochim Acta B At Spectrosc 66:193–241. doi:10.1016/j.sab.2010.12.011
De Corte F, Simonits A, Dewispelaere A et al (1989) k 0-measurements and related nuclear-data compilation for (n, gamma) reactor neutron-activation analysis. J Radioanal Nucl Chem 133(1):3–41
De Corte F, Simonits A (2003) Recommended nuclear data for use in the k(0) standardization of neutron activation analysis. At Data Nucl Data Tables 85(1):47–67
Simonits A, De Corte F, Hoste J (1976) Zirconium as a multi-isotopic flux ratio monitor and a single comparator in reactor-neutron activation-analysis. J Radioanal Chem 31(2):467–486
De Corte F, Moens L, Simonits A, de Wispelaere A, Hoste J (1979) Instantaneous alpha-determination without Cd-cover in the 1−E−1+alpha epithermal neutron-spectrum. J Radioanal Chem 52(2):295–304
El Nimr T, De Corte F, Moens L, Simonits A, Hoste J (1981) Epicadmium neutron-activation analysis (ENAA) based on the k 0-comparator method. J Radioanal Chem 67(2):421–435
Szőke R, Sziklai-László I (2008) Epiboron NAA: an option to analyze unfavorable matrices. J Radioanal Nucl Chem 275(1):89–95. doi:10.1007/s10967-007-6977-6
Basenko VK, Berlizov AN, Malyuk IA, Tryshyn VV (2005) NAAPRO: a code for predicting results and performance of neutron activation analysis. J Radioanal Nucl Chem 263(3):675–681
Kis Z, Völgyesi P, Szabó Z (2013) DÖME: revitalizing a low-background counting chamber and developing a radon-tight sample holder for gamma-ray spectroscopy measurements. J Radioanal Nucl Chem 298:2029–2035. doi:10.1007/s10967-013-2691-8
Fazekas B, Belgya T, Dabolczi L, Molnar G, Simonits A (1996) HYPERMET-PC: program for automatic analysis of complex gamma- ray spectra. J Trace Microprobe Tech 14(1):167–172
Simonits A, Ostor J, Kalvin S et al (2003) HyperLab: a new concept in gamma-ray spectrum analysis. J Radioanal Nucl Chem 257(3):589–595
Rausch H, Simonits A, Sziklai-László I (1994) A PC-program for element concentration calculations in neutron activation analysis. Izotóptechnika Diagnosztika 37:1–8 (in Hungarian)
Rossbach M, Blaauw M, Bacchi MA, Lin Xilei (2007) The k0-IAEA program. J Radioanal Nucl Chem 274(3):657–662
De Corte F, van Sluijs R, Simonits A et al (2001) Installation and calibration of Kayzero-assisted NAA in three Central European countries via a Copernicus project Appl. Radiat Isot 55(3):347–354
Blaauw M, Fleming RF, Keyser R (2001) Digital signal processing and zero-dead-time counting. J Radioanal Nucl Chem 248:309–313
Kennedy G, Tye P, St-Pierre J (2001) J Radioanal Nucl Chem 248:339–343
Pommé S (2001) Experimental test of the ‘‘zero dead time’’ count-loss correction method on a digital gamma-ray spectrometer. Nucl Instrum Method A 474:245–252
Kubesova M, Kucera J (2011) Comparison of Kayzero for Windows and k0-IAEA software packages for k0 standardization in neutron activation analysis. Nucl Instrum Method A 654:206–212
Vermaercke P, Robouch P, Eguskiza M, De Corte F, Kennedy G, Smodis B, Jacimovic R, Yonezawa C, Matsue H, Lin X, Blaauw M, Kucera J (2006) Characterisation of synthetic multi-element standards (SMELS) used for the validation of k 0-NAA. Nucl Instrum Methods A 564:675–682
Sziklai-László I, Gméling K (2013) Results of the k 0-based instrumental neutron activation analysis (k 0-INAA) for WEPAL plant and soil IPE/ISE 2013 in Hungary. In: Annex on CD-ROM of IAEA TECDOC. TECDOC and Annex connected to IAEA TC project RER1007: workshop on inter-comparison feedback of NAA and other analytical techniques proficiency tests performed in 2012–2013
IAEA Minor Actinide Neutron Reaction Data (MANREAD) Coordinated research project, 2006–2010. https://www-nds.iaea.org/manread/ Accessed on 04 Feb 2016
Szentmiklósi L, Révay Z, Belgya T et al (2008) Combining prompt gamma activation analysis and off-line counting. J Radioanal Nucl Chem 278(3):657–660
Acknowledgments
The authors are thankful to András Simonits, Rózsa Baranyai, as well as to the reactor staff for the continuous support, in particular Csaba Katona and József Janik. We also gratefully acknowledge the Infrastructure Upgrade Initiative of the Hungarian Academy of Sciences (MTA INFRA 2013 and 2015) for financing the technical upgrade of the laboratory. We thank Canberra-Packard Hungary Ltd. and Spektrum Méréstechnika Ltd. for borrowing us the digital gamma spectrometers for testing. Certain commercial equipment, instruments, or software are identified in this paper in order to specify the procedures in adequate detail. This identification does not imply recommendation or endorsement by the authors or by their research center, nor does it imply that the equipment or software identified are necessarily the best available for the purpose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Szentmiklósi, L., Párkányi, D. & Sziklai-László, I. Upgrade of the Budapest neutron activation analysis laboratory. J Radioanal Nucl Chem 309, 91–99 (2016). https://doi.org/10.1007/s10967-016-4776-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-016-4776-7