
Abstract
A fluorinated diamine, (2,5-bis(4-amino-3-trifluoromethylphenoxy)phenyl)diphenyl-phosphine oxide (2), was synthesized via Williamson reaction and hydrogenation. A series of polyimides with side diphenylphosphine oxide and trifluoromethyl groups were prepared by the high-temperature one-pot polymerization of diamine 2 with several commercial aromatic dianhydrides. The resulting PIs exhibit excellent solubility in common organic solvents (i.e., tetrahydrofuran, trichloromethane etc.) and are easily processed into light color transparent films (thickness: 20 ± 1 μm) through the blade-coating method. The transmittance of PI films is above 86% in the visible light region (400—760 nm). They also show good thermal stability with the glass transition temperatures from 246 to 286 °C. The limiting oxygen index values of them exceed 38.3%. At the same time, they display the low water absorption (0.89—1.32%) and good mechanical properties (tensile strength: 72.3—153.24 MPa, Young’s modulus: 1.4—2.3 GPa, elongation at break: 6.9—12.5%). They are promising candidates for advanced flame-retardant and optical film materials.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As a class of high performance materials, aromatic polyimides (PIs) have been widely applied in various high-tech fields due to their many advantages of outstanding chemical and thermal resistance, good dielectric properties, high mechanical properties and dimensional stability [1,2,3,4,5]. However, conventional aromatic PIs usually have darker color and poor processability due to the presence of charge transfer complexes (CTC) and rigid backbones. In order to improve the processability of PIs, various strategies have been developed, including the introduction of flexible linkages, aliphatic group, bulky side groups, fluorine-containing monomer, asymmetric monomer and so on into the molecular structure of PIs [6,7,8,9,10,11]. Especially, trifluoromethyls are often used to prepare fluorinated PIs with excellent optical transparency, solubility and processability because the low polarity of trifluoromethyls reduces the chain packing and the CTC formation of PIs. On the other hand, the introduction of trifluoromethyls into PIs sometimes brings some negative effects, such as poor adhesion, low glass-transition temperature (Tg), high coefficient of thermal expansion (CTE) and so on [12]. Unfortunately, there is a mutually restrictive relationship among the transparency, the thermal stability and the flame retardancy of PIs. The approaches to improve the optical transparency of PIs usually reduce their flame retardancy and thermal stability at a certain degree. This restricts the application of PIs in the fields of optics, aerospace or microelectronics [13,14,15].
In recent years, various structural units containing phosphorus has been introduced into PIs to improve their thermal oxidative stability, dielectric properties, optical transparency, radiation resistance etc. and facilitate their applications in optics, electronics, flame inhibition and aerospace fields [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. The PIs containing phenylphosphine oxide (PPO) groups are one kind of the outstanding representatives. Because of the large steric volume and the strong polar P = O bond of PPO, they demonstrated excellent optical transparency, adhesion properties and miscibility with many polymers [24]. For example, Yoon et al. synthesized some diamines containing fluorine and phosphine oxide moiety simultaneously to prepare PIs with excellent adhesion properties and thermal stability [24,25,26,27,28]. You et al. introduced phosphine oxide and thioether into the main chains of PIs and endowed them high refractive index for advanced optical applications [29]. Qiu and co-workers reported some co-PI fibers containing diphenylphosphine oxide groups, which exhibited good flame retardancy and high thermal stability [30, 31]. In addition, Yoon and Connell reported some novel dianhydride monomers with phosphine oxide moiety to prepare PIs for potential flexible printed circuits and space applications [32, 33]. Yang et al. reported some PIs containing PPO moiety were resistant to atomic oxygen erosion [34,35,36].
Here, we attempted to introduce diphenylphosphine oxide units as side group and trifluoromethyls at the ortho-position of amino group into PIs to improve their solubility, optical transparency and flame-retardancy without scarifying their thermal stability. So a novel aromatic diamine, (2,5-bis(4-amino-3-trifluoromethylphenoxy)phenyl)diphenyl-phosphine oxide (2), was first synthesized from 2,5-dihydroxyphenyl(diphenyl)phosphine oxide and 4-chloro-1-nitro-2-(trifluoromethyl)-benzene via Williamson reaction and then hydrogenation. Then a series of PIs were prepared from diamine 2 and various aromatic dianhydrides by the high-temperature one-step method. The various properties of PIs, especially the effects of side diphenylphosphine oxide units and trifluoromethyls at the ortho-position of amino group, were investigated in detail.
Experimental
Materials
2,5-dihydroxyphenyl(diphenyl)phosphine oxide and 4-chloro-1-nitro-2-trifluoromethylbenzene were purchased from TCI (Shanghai) Development Co., Ltd. and Adamas Reagent Co. Ltd. respectively. N,N-dimethylacetamide (DMAc) (Shanghai Titan Scientific Co. Ltd.) and m-cresol (Shanghai Macklin Biochemical Co. Ltd.) were purified by distillation prior to use. Pyromellitic dianhydride (PMDA, Sinopharm Chemical, China), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA, Adamas, China), 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (BTDA, J&K, China), 4,4′-oxydiphthalic anhydride (ODPA, Adamas, China), and 4,4′-hexafluoroisopropylidenediphthalic anhydride (6FDA, Shanghai Titan Scientific, China) were recrystallized from acetic anhydride prior to use. Commercially available palladium on active carbon (Pd/C, 10%) was purchased from Adamas Reagent Co. Ltd., potassium carbonate, toluene, N,N-dimethylformamide (DMF), ethanol, isoquinoline were purchased from Sinopharm Chemical Reagent Co. Ltd. and used as received.
Measurement
1H, 13C, 31P NMR spectra, 1H–1H correlated spectroscopy (COSY) and 1H–13C heteronuclear single quantum coherence (HSQC) spectra were recorded on a Bruker AM-500 (500 MHz) spectrometer with CDCl3 or DMSO-d6 as solvents. The mass spectral analyses of liquid chromatography-mass spectrometer and high-resolution mass spectrometer (HRMS) were performed on ACQUITYTM UPLC & Q-TOF MS Premier. The melt points were observed by the SGW X-4 melting-point apparatus with microscope. All other reagents were commercial products with analytical grade purity and used as received. Differential scanning calorimetry (DSC) curves were obtained on a TA Discovery DSC Q2000 thermal analyzer under N2 atmosphere at a heating rate of 20 K min–1 from 40 to 350 °C. Thermogravimetric analysis (TGA) was performed on a TA Discovery TGA Q5000 thermal analyzer under nitrogen at a heating rate of 20 K min–1 from 50 to 850 °C. FTIR measurement was obtained on a Perkin-Elmer Fourier transform infrared spectrometer. UV–vis spectra were recorded on a Perkin-Elmer Lambda 20 UV–vis spectrometer at room temperature. The mechanical property of the PI films was performed on an INSTRON 4465 tensile tester at a drawing rate of 2.0 mm min–1. Molecular weight carried out on Perkin-Elmer's 200 GPC analyzer (polystyrene calibration), using N,N-dimethylformamide as the eluent containing 0.02 M LiBr (0.6 mL min–1). Elemental analysis was measured with a Vario EL cube elemental analyzer. Inductive coupled plasma emission spectrometer (ICP) was measured with a Thermo Scientific™ iCAP 7600 ICP-OES Plasma spectrometer. The limiting oxygen index (LOI) values were measured on JF-3 oxygen index meter (Nanjing Jionglei Instrument Equipment Co., Ltd, China). The XRD measurement was carried out using Cu/K-α radiation and a Bruker D8 advanced X-ray diffractometer with a 2θ range of 5—50°. The water absorption rate was determined by a vacuum-dried film immersed in water at 25 °C for 24 h, and was calculated from the difference in weights. The CTE value of the PI samples (length 15 mm, width 6 mm, and greater than 20 μm thickness) were detected by the heating rate of 10 °C min–1 from 50 to 330 °C with a 0.02 N expansion force under nitrogen through thermomechanical analysis (TMA) on the Mettler Toledo TMA / SDTS841e thermal analyzer system.
Intermediate and monomer synthesis
10.00 g (32.23 mmol) 2,5-dihydroxyphenyl(diphenyl)phosphine oxide, 17.45 g (77.35 mmol) 4-chloro-1-nitro-2-trifluoromethylbenzene, 13.36 g (96.69 mmol) potassium carbonate, 250 ml DMF and 100 ml toluene were charged into a 500 mL three-neck flask fitted with a magnetic stirrer, a reflux condenser with Dean-Stark trap and a nitrogen inlet. The mixture was heated to 120 °C and kept at the reflux for 16 h to remove the by-product H2O. Then the temperature of the mixture was raised to 140 °C and kept the reaction until no by-product H2O was observed in Dean-Stark trap. The mixture was cooled to room temperature and slowly poured into the vigorous stirring hot water. Some gray solid was obtained after the suction filtration. Finally, it was purified by the recrystallization in the mixed solvent of ethanol/dichloromethane (2:1, v/v) twice to produce the target product of 1 12.43 g, yield: 56%. C32H19F6N2O7P; M.P.: 170.6—171.8 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.24 (d, 1H), 8.00 (d, 1H), 7.73—7.67 (m, 5H), 7.63 (dd, 1H), 7.56 (dd, 1H), 7.51—7.41 (m, 8H), 7.13 (dd, 1H), 7.02 (d, 1H); 31P NMR (200 MHz, DMSO-d6): δ 22.21; HRMS (ESI, m/z): [M + H]+ calculated for 689.0912, found 689.0962. The FTIR data and 13C NMR spectrum (Fig. S1) with corresponding data of intermediate 1 were shown in Supplementary Information.
12.43 g (18 mmol) 1 dissolved in 50 mL ethanol, 100 mg 10% Pd/carbon catalyst and a magnetic stir bar were added in a hydrogen reduction autoclave. The autoclave was filled with 5 Mpa hydrogen and the suspension was stirred at 50 °C until the hydrogen pressure no longer decreased (about 1—2 days). Then the autoclave was cooled down to room temperature and the hydrogen pressure was released in the fume hood. 20 mL ethanol was added into the autoclave to dilute the suspension. The Pd/carbon catalyst was removed by the filtration with diatomite/filter paper and washed twice with ethanol (2 × 10 mL). The solvent was removed by rotary evaporation to produce the crude product of 2. It was purified by recrystallization twice in ethanol to provide some white crystals of diamine 2 10.43 g, yield: 92%. C32H23F6N2O3P; M.P.: 98.3—99.7 °C, 1H NMR (500 MHz, DMSO-d6): δ 7.70—7.66 (m, 4H), 7.57—7.54 (m, 2H), 7.50—7.47 (m, 4H), 7.22—7.19 (dd, J 1 = 13.5 Hz, J 2 = 3.0 Hz, 1H), 7.14—7.12 (dd, J 1 = 8.5 Hz, J 2 = 3.0 Hz, 1H), 7.10—7.08 (dd, J 1 = 9.0 Hz, J 2 = 3.0 Hz, 1H), 7.06—7.05 (d, J = 2.5 Hz, 1H), 6.90—6.88 (d, J = 8.5 Hz, 1H), 6.77—6.70 (m, 3H), 6.43—6.42 (d, J = 2.5 Hz, 1H), 5.55 (s, 2H), 5.49 (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ 154.18, 153.18, 144.80, 143.84, 143.34, 143.32, 132.46 (d, J = 106.0 Hz), 131.80, 131.33, 128.44, 125.57, 125.40, 124.49 (q, J = 270.4 Hz), 124.31 (q, J = 270.5 Hz), 123.59, 123.19 (d, J = 98.6 Hz), 122.84, 121.40, 118.68, 118.20, 117.85, 117.20 (q, J = 5.0 Hz), 116.88 (q, J = 5.0 Hz), 111.01 (q, J = 29.9 Hz), 110.80 (q, J = 29.9 Hz); 31P NMR (200 MHz, DMSO-d6): δ 23.50; HRMS (ESI, m/z): [M + H]+ calculated for 629.1429, found 629.1413.
Polymer synthesis
All the polyimides were synthesized via a high-temperature one-pot polycondensation and a typical procedure exhibited as follows (e.g., PI-2). 1 g (1.59 mmol) diamine 2, 0.707 g (1.59 mmol) 6FDA, 10 mL m-cresol and three drops isoquinoline were added into a 100 mL three-necked flask equipped with a mechanical stirrer under N2 atmosphere. The mixture was stirred and slowly heated to 50 °C until the solid dissolved completely to form a clear solution in 8 h. Then the temperature of the solution was processed with the following heating program, at 80 °C for 8 h, 100 °C for 8 h, 120 °C for 10 h, 150 °C for 10 h, and 180 °C for 10 h. After cooling to room temperature, 5 mL chloroform was added to dilute the yellow viscous mixture under the stirring. Then the diluted yellow mixture was slowly poured into 500 mL vigorously stirred methanol to give some white fiber-like precipitate. The precipitate was collected by filtration and dried at 100 °C in vacuum overnight to afford PI-2. For further purification, PI-2 was reprecipitated twice from CH3Cl into methanol.
PI-2 (2-6FDA)
Yield: 93%. FTIR (blade-coating film, cm–1): ν = 1792 (C = O), 1738 (C = O), 1620, 1591, 1505, 1472, 1435, 1381 (C–N), 1316, 1256, 1212 (P = O), 1172, 1146, 1103, 1048, 985, 925, 891, 847, 724, 710, 545. 1H NMR (500 MHz, CDCl3): δ 8.06—8.03 (m, 2H), 8.00—7.96 (m, 2H), 7.89—7.86 (m, 2H), 7.83—7.80 (m, 1H), 7.76—7,72 (m, 4H), 7.52—7.49 (m, 2H), 7.47 (s, 1H), 7.41—7.38 (m, 4H), 7.35—7.29 (m, 3H), 7.12—7.06 (m, 2H), 6.87—6.85 (m, 2H).
PI-1 (2-PMDA)
Yield: 91%. FTIR (blade-coating film, cm–1): ν = 1785 (C = O), 1738 (C = O), 1618, 1506, 1472, 1435, 1376 (C–N), 1316, 1253, 1218 (P = O), 1170, 1141, 1104, 1046, 925, 821, 728, 695, 545. 1H NMR (500 MHz, CDCl3): δ 8.52—8.49 (m, 2H), 7.82 (d, 1H), 7.77—7.73 (m, 4H), 7.54—7.51 (m, 2H), 7.48 (s, 1H), 7.42—7.40 (m, 4H), 7.36—7.31 (m, 3H), 7.13—7.09 (m, 3H), 6.89 (d, 1H), 6.86 (s, 1H).
PI-3 (2-BTDA)
Yield: 93%. FTIR (blade-coating film, cm–1): ν = 1785 (C = O), 1735 (C = O), 1678, 1619, 1505, 1472, 1434, 1381 (C–N), 1315, 1250, 1218 (P = O), 1175, 1139, 1101, 1048, 925, 877, 726, 705, 544. 1H NMR (500 MHz, CDCl3): δ 8.32—8.27 (m, 4H), 8.16—8.13 (m, 2H), 7.81(d, 1H), 7.76—7.72 (m, 4H), 7.53—7.50 (m, 2H), 7.47 (s, 1H), 7.41—7.30 (m, 7H), 7.11—7.09 (m, 2H), 6.88—6.85 (m, 2H).
PI-4 (2-BPDA)
Yield: 90%. FTIR (blade-coating film, cm–1): ν = 1782 (C = O), 1731 (C = O), 1618, 1505, 1472, 1434, 1380 (C–N), 1315, 1252, 1225 (P = O), 1175, 1139, 1101, 1048, 925, 891, 844, 744, 727, 695, 545. 1H NMR (500 MHz, CDCl3): δ 8.25—8.23 (m, 2H), 8.13—8.06 (m, 4H), 7.84—7.81 (m, 1H), 7.78—7.74 (m, 4H), 7.55—7.52 (m, 2H), 7.48 (s, 1H), 7.45—7.38 (m, 4H), 7.36—7.27 (m, 3H), 7.14—7.06 (m, 2H), 6.89—6.87 (m, 2H).
PI-5 (2-ODPA)
Yield: 91%. FTIR (blade-coating film, cm–1): ν = 1785 (C = O), 1732 (C = O), 1611, 1505, 1474, 1434, 1381 (C–N), 1315, 1276, 1247, 1218 (P = O), 1175, 1138, 1100, 1047, 925, 888, 845, 818, 749, 728, 695, 544. 1H NMR (500 MHz, CDCl3): δ 8.03—8.00 (m, 2H), 7.83—7.80 (m, 1H), 7.76—7.72 (m, 4H), 7.58—7.56 (m, 2H), 7.51—7.50 (m, 4H), 7.45 (s, 1H), 7.41—7.38 (m, 4H), 7.34—7.28 (m, 3H), 7.10—7.05 (m, 2H), 6.87—6.84 (m, 2H).
Film preparation
The blade-coating method was adopted to prepare PI films. A typical procedure to prepare PI-2 film (Thickness: 20 ± 1 μm) was as follows: 1.2 g PI-2 was dissolved in 10 mL DMAc to give a transparent solution and then kept at 60 °C for 8 h. The solution was filtered through a 0.7 μm fiberglass syringe filter and then blade-coated on a clean glass substrate at 60 °C by using Elcometer 4340 Automatic Film Applicator. The height of the blade and the coating speed were set to 80 μm and 0.2 inches s–1, respectively. The solution on the glass substrate was maintained at 60 °C for 4 h to remove the solvent and form a solid film. Then the film on the glass substrate was dried in vacuum at 100 °C for 6 h and 200 °C for 12 h to remove residual solvent. After cooling to room temperature, the glass substrate was immersed in deionized water to take off the PI film.
Results and discussion
The novel diamine 2 with side diphenylphosphine oxide units and trifluoromethyls at the ortho-position of amino group was synthesized in two steps as shown in Scheme 1. In the first step, the water content in the reaction system is the key factor to affect the yield of Williamson reaction between 4-chloro-1-nitro- 2-trifluoromethylbenzene and 2,5-dihydroxyphenyl(diphenyl)phosphine oxide. Williamson reaction usually can be proceeded smoothly at high temperature with K2CO3 as a weak base for deprotonation. According to the previous report, in the nucleophilic displacement reaction of 2-(6-oxido-6H-dibenz < c,e > < 1,2 > oxaphosphorin-6-yl)-1,4-benzenediol and 4-fluoronitrobenznene at high temperature, the P–C bond in side group was cleaved easily in the presence of H2O and K2CO3 to decrease the yield of target product [37]. Although cesium fluoride used as a base can avoid the above problem, hypertoxic hydrogen fluoride would be generated [20]. Herein, toluene was added into Williamson reaction system as an azeotropic dehydrating agent to remove the water by reflux at 120 °C and decrease the cleavage of P–C bond in side group of 2,5-dihydroxyphenyl(diphenyl)phosphine oxide. Finally, the reaction temperature was raised to 140 °C to complete Williamson reaction until no by-product H2O was found in Dean-Stark trap. Cooled to room temperature, the reaction mixture was slowly poured into the vigorous stirring hot water to precipitate some gray solid of dinitro-intermediate 1. It was purified by the recrystallization in the mixed solvent of ethanol/dichloromethane to afford some beige crystals with the yield of 56%. In the second step, dinitro-intermediate 1 was transformed into diamine 2 by the conventional hydrogenation reaction with Pd/C as the catalyst in ethanol. It was purified by recrystallization twice in ethanol to afford some white crystals with the yield of 92%.

Synthesis of diamine 2
The chemical structure of diamine 2 was confirmed by various spectroscopic measurements. The molecular mass of diamine 2 was 629.1413 measured by HRMS, which was in good agreement with the calculated value of 629.1429. The 1H NMR spectrum of diamine 2 was shown in Fig. 1a) and all the signals were labeled by numbers. The proton signals (H10/H10') of two amino groups displayed distinct absorptions at 5.49 and 5.55 ppm, respectively, which is ascribed to its asymmetric structure. Similarly, the proton pairs (H7/H7', H8/H8', and H9/H9') located in the amino-substituted benzenes also exhibited separate signals at 6.42—7.10 ppm. Among them, the proton signals (H8' and H9') were overlapped with the one (H5) belong to another phenyl ring. The asymmetric chemical structure of diamine 2 was also verified by the 13C NMR spectrum in Fig. 1b) and all the signals were labeled by letters. All the carbon atom signals of amino-substituted benzenes appeared at different chemical shift, but very close ones. The carbon signals of –CF3 (Cq/Cq'), –C–CF3 (Co/Co'), or –C–C–CF3 (Cp/Cp') exhibited the typical quartet absorptions at 100—120 ppm, and their coupling constants (1JC–F = 270.5 Hz, 2JCC–F = 29.9 Hz, 3JCCC–F = 5 Hz) were decreased sequentially due to the different coupling of C with F atoms in diamine 2. The carbons (Ca and Ce) in the diamine 2 exhibited clear double signals at 132.0—132.9 and 122.8—123.6 ppm due to the coupling of C with P atoms. However, they exhibited different coupling constants of JC–P = 106.0 Hz (Ca) and JC–P = 98.6 Hz (Ce), which confirmed diphenylphosphine oxide moiety was located as side group. Furthermore, the quartet and doublet of carbon splitted by 19F and 31P was labelled by the capital letters of F and P respectively, in Fig. 1b. The 31P NMR spectrum of diamine 2 (Fig. 1c) exhibited only one sharp and strong signal at 23.5 ppm, which indicated that only one kind of P atom in diamine 2.

a) 1H, b) 13C and c) 31P NMR spectra of diamine 2
Due to the structural asymmetry of diamine 2, its 1H and 13C NMR spectra were complicated, including signal overlapping and difficulty to determine C–H correlation etc. Therefore, 1H–1H COSY and 1H–13C HSQC spectra were adopted to further confirm the chemical structure of diamine 2. As shown in Fig. 2a), the aromatic proton signals in the same benzene can be correlated and distinguished with each other correctly in 1H–1H COSY spectrum. The 1H–13C HSQC spectrum of diamine 2 was shown in Fig. 2b). The prominent correlations of Ch/H5, Cl'/H8' and Cm'/H9' were observed at δC/δH 6.8/117.8, 6.8/118.2 and 6.7/125.4 ppm to distinguish H5, H8' and H9' easily, which were overlapped heavily in 1H NMR spectrum. As a result, the 1D and 2D NMR spectra confirmed that the diamine 2 was in accordance with the expected structure.

a) 1H–1H COSY and b) 1H–13C HSQC spectra of diamine 2
The preparation process of the PIs from diamine 2 and various dianhydrides was shown in Scheme 2. Due to the introduction of trifluoromethyl into the ortho-position of amino group and causing steric hindrance, the reactivity of diamine 2 was decreased relatively. So the high-temperature one-step method was adopted to prepare all PIs in m-cresol with isoquinoline as the catalyst and the solid content of 10 wt%.

Synthesis of PIs from diamine 2 with various dianhydrides
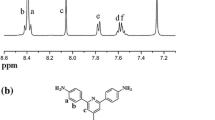
The resulting PIs were characterized by 1H NMR, FT-IR, elemental analysis, and GPC measurements. The 1H NMR spectra of all PIs were shown in Fig. 3. All the signals of each PI were labeled by numbers and assigned to the corresponding protons. In detail, the proton signals at the upfield region of 6.8—7.8 ppm were ascribed to the diamine unit with strong absorptions, while those at the downfield region of 7.9—8.5 ppm were attributed to the dianhydride unit with clear absorptions. Among them, only for PI-5, the proton signals (H10 and H11) close to ether bond in ODPA unit were embedded in the region of the diamine unit due to the electron-donating effect of ether bond.

1H NMR spectra of PIs
The FTIR spectra of the PIs were shown in Fig. 4. The characteristic absorption bands of imide units were clearly observed at 1778—1787 cm–1 (C = O asymmetrical stretching), 1725—1730 cm–1 (C = O symmetrical stretching), 1367—1374 cm–1 (C–N stretching vibration) and 721 cm–1 (imide ring deformation). In addition, the strong absorption bonds of C–F were found at 1000—1200 cm–1. The characteristic absorption bonds of P = O and P–Ph were also found around 1220 cm–1 and 1620 cm–1, respectively. No obvious absorption bonds of amide units were observed in the region between 3220 and 3440 cm−1 (N–H stretching), which indicated the imidization of all PIs were completely.

FTIR spectra of PIs
The elemental analysis data of all PIs were shown in Table 1. Due to phosphorus element not in the inspection range of the organic element analyzer, the content of phosphorus element was measured by the ICP. All above results were in good agreement with the theoretically calculated values and further confirmed the chemical structures of them.
The molecular weights of all PIs were measured by GPC and the results were shown in Table 2. Their number-average molecular weights and molecular weight distribution were in the range from 1.40 to 4.39 × 104 and 1.76 to 3.22, respectively. The water absorption rates of all PI films were measured at 25 °C and the results were listed in Table 2. Compared with the water absorption rate of 3.1% for commercial Kapton film [38], the water absorption rates of these PI films were much lower and in the range from 0.89% to 1.32%. Figure 5 presents the contact angle measurement photos of water droplet on the PI film surface. The contact angles of these PI films were around 90°. Among them, PI-1 and PI-5 exhibited a weak hydrophilicity because their contact angles were lower than 90° (87.3° and 89.0°, respectively). On the contrary, PI-2, PI-3 and PI-4 displayed a somewhat hydrophobicity because their contact angles were slightly larger than 90°.

The contact angles (CA) of a water droplet on the surface of PI films
The solubility of all PIs was examined by dissolving 10 mg polymer in 1 mL solvent at room temperature and the results were listed in Table 4. All of them exhibited good solubility not only in polar aprotic solvents (e.g., DMF, DMSO and NMP), but also in common low boiling solvents (e.g., CH2Cl2, CHCl3 and THF). This can be ascribed to the introduction of bulky diphenylphosphine oxide and − CF3 groups into PIs significantly restrained the formation of CTC while the steric hindrance of bulky side groups reducing the stacking density of PI chains. Their synergistic effect improved the solubility of the PIs. Such good solubility of these PIs made them simply process into light-colored or colorless transparent films by the blade coating of polymer solution.
The thermal properties of the PIs were evaluated by TGA, DSC and TMA measurements under N2 atmosphere. Their TGA curves were shown in Fig. 6b) and the values of T5wt% were summarized in Table 3. All PIs exhibited high thermal stability and kept their weight loss within 5% up to 482 °C. The major thermal weight loss of them occurred in the temperature range from 580 to 610 °C, which was attributed to the thermal degradation of PI backbones. The residual carbon weights of all PIs were higher than 49.9% at 850 °C. The glass transition temperatures (Tgs) of all PIs were measured by DSC and TMA measurements. The resulting Tgs were summarized in Table 2 and the DSC curves were shown in Fig. 6a). The Tgs of these PIs were in the range from 246 to 286 °C by DSC measurements as well as from 265 to 310 °C by TMA measurements, which were decreased with decreasing rigidity of dianhydrides in the order of PMDA > BPDA > 6FDA > BTDA > ODPA. For example, PI-5 possessed the lowest Tg (246 °C) due to the flexible ether linkage in ODPA, whereas PI-1 displayed the highest Tg (286 °C) because of the rigid structure of PMDA. The Tgs measured by TMA measurements exhibited the same trend as those by DSC measurements, but they were about 20 °C higher than those by DSC measurements on average. The Tgs of PIs containing diphenylphosphine oxide and –CF3 groups in previous studies were also in the similar temperature range from 200 to 310 °C [15, 20, 24,25,26,27]. The PIs made from a similar diamine (BATFDPO, –CF3 at the meta-position of amino group) exhibited very similar thermal stability (e.g., Tgs: 244 – 266 °C, T5wt%: 472 – 477 °C) as ours, which means the substitution location of –CF3 had little effect on the thermal stability of PIs [20]. However, the PIs synthesized from a diamine with diphenylphosphine oxide in the main chain displayed relatively low thermal stability (e.g., Tgs: 200 – 232 °C, T5wt%: 441 – 487 °C) comparing with ours, which indicated that diphenylphosphine oxide as the side group was beneficial to improve the thermal stability of PIs [15].

a) DSC curves of PIs; b) TGA curves of PIs; c) UV–vis spectra of PI films (Thickness: 20 ± 1 μm); d) X-ray diffraction curves of PI films
Generally, the LOI values of PIs can be calculated according to van Krevelen equation [39]:
Here, CR is the weight percentage of char residue for PI at 850 °C.
The theoretical calculation LOI values of our PI films were shown in Table 3 and they were in the range from 37.5% to 40.5%. We also measured the LOI values of our PI films by oxygen index meter and the results were in the range of 38.3—41.0%, which were slightly higher than the theoretical calculation ones. The calculated LOI values and the measured ones of our PIs were higher than those of previously reported PIs without PPO moieties. For example, the LOI value of ODA-BPDA is 34.9% while the LOI value of PI-4 is 40.5% (measured) and 39.1% (calculated) [20]. These results indicated that the introduction of PPO moieties into PIs could improve their flame retardance properties.
The UV–vis spectra of all PI films (Thickness: 20 ± 1 μm) were displayed in Fig. 6c) and the corresponding optical properties were listed in Table 5. The transmittance of all PI films were above 85% in the visible light range (400—760 nm) and their cut-off wavelengths were in the range from 308 to 357 nm. Among them, PI-2 film prepared from diamine 2 and 6FDA had the highest average transmittance of 92% and PI-3 prepared from diamine 2 and BTDA had the lowest cut-off wavelength of 308 nm. This indicated that it was the effective approach to increase the optical transmittance of the PI films by introducing both PPO moieties and –CF3 group. These PI films had lower UV cutoff wavelengths than those of PIs with similar structures (λ0: 350—370 nm [15] or 312—368 nm [20]).
The morphology of all PI films was investigated by X-ray diffraction (XRD) measurements and the corresponding diffraction patterns were shown in Fig. 6d). Obviously, all the PI films were in amorphous nature with some broad diffusional peaks, which indicated the side diphenylphosphine oxide and –CF3 groups disturbed the packing of PI chains and decreased their packing density. The d-spacings of these PIs were calculated on the base of Bragg’s equation and the results were in the range of 6.56—6.61 Å. Comparing with the d-spacing of 4.78 Å for Kapton, the d-spacings can be improved by introducing side diphenylphosphine oxide and –CF3 groups into PIs [40].
The mechanical properties of these PI films were measured by tensile tests at 25 °C and the results were listed in Table 6. The values of tensile strength, tensile modulus, and elongation at break were in the range of 72.3—153.24 MPa, 1.4—2.3 GPa and 6.9—12.5%. The dimensional stability of the PI films was measured by TMA measurement and the CTE values were summarized in Table 6. Generally, the CTE values have a major relationship with the linear/stiff backbone structure of PIs and the level of PI chain alignment to the film in-plane orientation [41, 42]. Here these PI films exhibited the relatively high CTE values in range from 44 to 69 ppm °C–1, which was attributed to the introduction of side diphenylphosphine oxide and –CF3 groups increasing the d-spacings as well as free volumes. Among them, PI-4 (2-BPDA) possessed the lowest CTE (44 ppm °C–1) due to containing the relatively rigid dianhydride BPDA, while PI-2 (2-6FPA) had the largest CTE (69 ppm °C–1) due to the relatively flexible dianhydride 6FDA.
Conclusion
A series of PIs containing side diphenylphosphine oxide and –CF3 groups were synthesized by the high-temperature one-pot mothed. All of them exhibit excellent solubility and can be process into transparent and tough films by the blade coating method. The transmittance of the PI films (20 ± 1 μm) is above 86% in the region from 400 to 760 nm. They also show good thermal stability (T5wt% > 482 °C) with the Tgs from 246 to 286 °C. The LOI values of them exceed 38.3%, which means they have good flame retardancy. Meanwhile they have good mechanical properties (tensile strength: 72.3—153.24 MPa, Young’s modulus: 1.4—2.3 GPa, elongation at break: 6.9—12.5%) with the low water absorption (0.89—1.32%). In summary, it is a practical and efficient approach to improve the transparency and flame-retardancy of PI films by introducing –CF3 and side diphenylphosphine oxide groups into PIs. They are promising candidates for advanced flame-retardant and optical film materials.
References
Tapaswi PK, Ha CS (2019) Recent trends on transparent colorless polyimides with balanced thermal and optical properties: design and synthesis. Macromol Chem Phys 220:1800313. https://doi.org/10.1002/macp.201800313
Choi MC, Kim Y, Ha CS (2008) Polymers for flexible displays: From material selection to device applications. Prog Polym Sci 33:581–630. https://doi.org/10.1016/j.progpolymsci.2007.11.004
Zhuang Y, Seong JG, Lee YM (2019) Polyimides containing aliphatic/alicyclic segments in the main chains. Prog Polym Sci 92:35–88. https://doi.org/10.1016/j.progpolymsci.2019.01.004
Yi L, Huang W, Yan DY (2017) Polyimides with side groups: synthesis and effects of side groups on their properties. J Polym Sci Part A: Polym Chem 55:533–559. https://doi.org/10.1002/pola.28409
Hergenrother PM (2016) The use, design, synthesis, and properties of high performance/high temperature polymers: an overview. High Perform Polym 15:3–45. https://doi.org/10.1177/095400830301500101
Ma H, Jen AY, Dalton LR (2002) Polymer-based optical waveguides: materials, processing, and devices. Adv Mater 14:1339–1365. https://doi.org/10.1002/1521-4095(20021002)14:19%3c1339::AID-ADMA1339%3e3.0.CO;2-O
Ogbonna VE, Popoola API, Popoola OM, Adeosun SO (2022) A review on polyimide reinforced nanocomposites for mechanical, thermal, and electrical insulation application: challenges and recommendations for future improvement. Polym Bull 79:663–695. https://doi.org/10.1007/s00289-020-03487-8
Maier G (2001) Low dielectric constant polymers for microelectronics. Prog Polym Sci 26:3–65. https://doi.org/10.1016/S0079-6700(00)00043-5
Gouzman I, Grossman E, Verker R, Atar N, Bolker A, Eliaz N (2019) Advances in polyimide-based materials for space applications. Adv Mater 31:1807738. https://doi.org/10.1002/adma.201807738
Ni HJ, Liu JG, Wang ZH, Yang SY (2015) A review on colorless and optically transparent polyimide films: Chemistry, process and engineering applications. J Ind Eng Chem 28:16–27. https://doi.org/10.1016/j.jiec.2015.03.013
Wu XM, Shu C, He XQ, Wang SB, Fan X, Yu ZH, Yan DY, Huang W (2020) Optically transparent and thermal-stable polyimide films derived from a semi-aliphatic diamine: synthesis and properties. Macromol Chem Phys 221:1900506. https://doi.org/10.1002/macp.201900506
Sasaki S, Nishi S (1996) In Ghosh MK, Mittal KL (eds) Polyimides: Fundamentals and Applications, 1st edn. Marcel Dekker, NewYork. Chapter 4, 71–120. https://doi.org/10.1201/9780203742945
Hsiao SH, Lin KH (2005) Polyimides derived from novel asymmetric ether diamine. J Polym Sci Part A: Polym Chem 43:331–341. https://doi.org/10.1002/pola.20505
Choi WS, Harris F (2000) Synthesis and polymerization of trifluorovinylether-terminated imide oligomers. I Polymer 41:6213–6221. https://doi.org/10.1016/S0032-3861(99)00857-5
Zhu Y, Zhao P, Cai X, Meng WD, Qing FL (2007) Synthesis and characterization of novel fluorinated polyimides derived from bis[4-(4’-aminophenoxy)phenyl]-3,5-bis(trifluoromethyl)phenyl phosphine oxide. Polymer 48:3116–3124. https://doi.org/10.1016/j.polymer.2007.03.057
Wang D, Yu JJ, Duan GG, Liu KM, Hou HQ (2020) Electrospun polyimide nonwovens with enhanced mechanical and thermal properties by addition of trace plasticizer. J Mater Sci 55:5667–5679. https://doi.org/10.1007/s10853-020-04402-2
Wu L, Wu X, Qi HR, An YC, Jia YJ, Zhang Y, Zhi XX, Liu JG (2021) Colorless and transparent semi-alicyclic polyimide films with intrinsic flame retardancy based on alicyclic dianhydrides and aromatic phosphorous-containing diamine: Preparation and properties. Polym Adv Technol 32:1061–1074. https://doi.org/10.1002/pat.5153
Zhao Y, Gao H, Li GM, Liu FF, Dai XM, Dong ZX, Qiu XP (2018) Synthesis and AO resistant properties of novel polyimide fibers containing phenylphosphine oxide groups in Main Chain. Chin J Polym Sci 37:59–67. https://doi.org/10.1007/s10118-019-2179-2
Ni HJ, Xing Y, Dai XX, Zhang DJ, Li J, Liu JG, Yang SY, Chen XB (2020) Intrinsically heat-sealable polyimide films with atomic oxygen resistance: Synthesis and characterization. High Perform Polym 32:902–913. https://doi.org/10.1177/0954008320908652
Li Z, Liu JG, Gao ZQ, Yin ZH, Fan L, Yang SY (2009) Organo-soluble and transparent polyimides containing phenylphosphine oxide and trifluoromethyl moiety: Synthesis and characterization. Eur Polym J 45:1139–1148. https://doi.org/10.1016/j.eurpolymj.2009.01.017
Wu BH, Zhang Y, Yang DY, Yang YB, Yu Q, Che L, Liu JG (2019) Self-healing anti-atomic-oxygen phosphorus-containing polyimide film via molecular level incorporation of nanocage trisilanolphenyl POSS: preparation and characterization. Polymers 11:1013. https://doi.org/10.3390/polym11061013
Xu RX, Ma JL, Zhou R, Sun HJ, Xu DW, Zeng Z (2020) Black phosphorus nanoflakes/polyimide composite films with excellent dielectric and mechanical properties. J Mater Sci: Mater El 31:3303–3311. https://doi.org/10.1007/s10854-020-02878-x
Schartel B (2010) Phosphorus-based flame retardancy mechanisms-old hat or a starting point for future development?. Materials 3:4710–4745. https://doi.org/10.3390/ma3104710
Jeong KU, Kim JJ, Yoon TH (2001) Synthesis and characterization of novel polyimides containing fluorine and phosphine oxide moieties. Polymer 42:6019–6030. https://doi.org/10.1016/S0032-3861(01)00012-X
Jeong KU, Young JJ, Yoon TH (2001) Synthesis and characterization of novel polyimide from bis-(3-aminophenyl)-4-(trifluoromethyl)phenyl phosphine oxide. J Polym Sci Part A: Polym Chem 39:3335–3347. https://doi.org/10.1002/pola.1316
Myung BY, Ahn CJ, Yoon TH (2005) Adhesion property of novel polyimides with 1-[3’,5’-bis(trifluoromethyl)phenyl] pyromellitic dianhydride. J Appl Polym Sci 96:1801–1809. https://doi.org/10.1002/app.21620
Lee CW, Kwak SM, Yoon TH (2006) Synthesis and characterization of polyimides from bis(3-aminophenyl)-2,3,5,6-tetrafluoro-4-trifluoromethylphenyl phosphine oxide (mDA7FPPO). Polymer 47:4140–4147. https://doi.org/10.1016/j.polymer.2006.03.002
Kwak SM, Yeon JH, Yoon TH (2006) Synthesis and characterization of polyimides from bis(3-aminophenyl)-4-(1-adamantyl)phenoxyphenyl phosphine oxide. J Polym Sci Part A: Polym Chem 44:2567–2578. https://doi.org/10.1002/pola.21364
Kim H, Ku BC, Goh M, Yeo H, Ko HC, You NH (2018) Synthesis and characterization of phosphorus- and sulfur-containing aromatic polyimides for high refractive index. Polymer 136:143–148. https://doi.org/10.1016/j.polymer.2017.12.052
Zhao Y, Li GM, Liu FF, Dai XM, Dong ZX, Qiu XP (2017) Synthesis and properties of novel polyimide fibers containing phosphorus groups in the side Chain (DATPPO). Chin J Polym Sci 35:372–385. https://doi.org/10.1007/s10118-017-1896-7
Zhao Y, Feng T, Li GM, Liu FF, Dai XM, Dong ZX, Qiu XP (2016) Synthesis and properties of novel polyimide fibers containing phosphorus groups in the main chain. RSC Adv 6:42482–42494. https://doi.org/10.1039/c6ra02344d
Thompson CM, Smith JG Jr, Connell JW (2003) Polyimides prepared from 4, 4’-(2-diphenylphosphinyl-1,4-phenylenedioxy)diphthalic anhydride for potential space applications. High Perform Polym 15:181–195. https://doi.org/10.1177/0954008303015002003
Bae YU, Yoon TH (2012) Synthesis and characterization of polyimides from 4-(diphenyl phosphine oxide)phenyl pyrromellitic dianhydride. J Appl Polym Sci 123:3298–3308. https://doi.org/10.1002/app.34934
Li Z, Song HW, He MH, Liu JG, Yang SY (2012) Atomic oxygen-resistant and transparent polyimide coatings from [3,5-bis(3-aminophenoxy)phenyl]diphenylphosphine oxide and aromatic dianhydrides: Preparation and characterization. Prog Org Coat 75:49–58. https://doi.org/10.1016/j.porgcoat.2012.03.007
Connell JW, Watson KA (2001) Space environmentally stable polyimides and copolyimides derived from bis(3-aminophenyl)-3,5-di(trifluoromethyl) phenylphosphine oxide. High Perform Polym 13:23–34. https://doi.org/10.1088/0954-0083/13/1/303
Reddy MR (1995) Effect of low earth orbit atomic oxygen on spacecraft materials. J Mater Sci 30:281–307. https://doi.org/10.1007/BF00354389
Liou GS, Hsiao SH (2001) Unexpected discovery of the formation of high-molecular-weight aromatic polyamides from unstoichiometric diacyl chloride/diamine components. High Perform Polym 13:S137–S152. https://doi.org/10.1088/0954-0083/13/2/313
Li SZ, Chen RS, Greenbaum SG (1995) NMR studies of water in polyimide films. J Polym Sci Part B: Polym Phys 33:403–409. https://doi.org/10.1002/polb.1995.090330308
Van Krevelen DW (1975) Some basic aspects of flame resistance of polymeric materials. Polymer 16:615–620. https://doi.org/10.1016/0032-3861(75)90157-3
Liu YW, Tang A, Tan JH, Chen CL, Wu D, Zhang HL (2021) Structure and gas barrier properties of polyimide containing a rigid planar fluorene moiety and an amide group: insights from molecular simulations. ACS Omega 6:4273–4281. https://doi.org/10.1021/acsomega.0c05278
Ishii J, Shimizu N, Ishihara N, Ikeda Y, Sensui N, Matano T, Hasegawa M (2010) Spontaneous molecular orientation of polyimides induced by thermal imidization (4): Casting- and melt-induced in-plane orientation. Eur Polym J 46:69–80. https://doi.org/10.1016/j.eurpolymj.2009.09.002
Hasegawa M, Matano T, Shindo Y, Sugimura T (1996) Spontaneous molecular orientation of polyimides induced by thermal imidization. 2. In-plane orientation Macromolecules 29:7897–7909. https://doi.org/10.1021/ma960018n
Acknowledgements
This work is supported by Equipment Research and Development Sharing Technology Project (No. 41421060301).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shu, C., Wu, X., Zhong, M. et al. Synthesis and properties of polyimides from a diamine containing side diphenylphosphine oxide and trifluoromethyl groups. J Polym Res 29, 394 (2022). https://doi.org/10.1007/s10965-022-02998-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-022-02998-4