
Abstract
A novel diamine 4,4′-(3-(tert-butyl)-4-aminophenoxy)diphenyl ether (4) was synthesized from 2-tert-butylaniline and 4,4′-oxydiphenol through iodination, acetyl protection, coupling reaction and deacetylation protection. Then some polyimides (PIs) were obtained by one-pot polycondensation of diamne 4 with several commercial aromatic dianhydrides respectively. They all exhibit enhanced solubility in organic solvents (such as NMP, DMF, THF and CHCl3 etc.) at room temperature. Their number-average molecular weights are in the range of (2.1–3.7) × 104 g/mol with PDI from 2.25 to 2.74 by GPC. They can form transparent, tough and flexible films by solution-casting. The light transparency of them is higher than 90% in the visible light range from 400 nm to 760 nm and the cut-off wavelengths of UV–vis absorption are below 370 nm. They also display the outstanding thermal stability with the 5% weight loss temperature from 525 °C to 529 °C in nitrogen atmosphere. The glass transition temperatures (T g s) are higher than 264 °C by DSC. XRD results demonstrate that these PIs are amorphous polymers with the lower water absorption (<0.66%). In summary, the incorporation of tert-butyl groups and multiple phenoxy units into the rigid PI backbones can endow them excellent solubility and transparency with relatively high T g s.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As one of famous high performance polymers, aromatic polyimides are widely used in many high-tech fields, such as automobile, membrane separation, adhesives, aerospace and micro-electronics etc. [1–6]. Due to the rigid backbones of traditional polyimides, most of them are difficult to process in solution or melting state, which limits their wide applications [7]. So many attentions have been focused on design and synthesis of PIs with excellent processability without sacrificing their inherent properties. Up to date, there are two main approaches to improve their processability. One is to improve their solubility by using aromatic monomers with asymmetric units, bulky side-groups or fluorine atoms [8–15]. The other is to improve their thermoplasticity by introducing flexible linkages into their backbone [16–20]. For example, Tamai et al. synthesized a series of PIs based on aromatic ether diamines having different numbers of benzene rings and found the T g s of these PIs decrease with increasing numbers of benzene rings in diamines. In other words, the thermostability of PIs decrease with increasing the number of flexible linkages in backbone. On the other hand, our previous studies demonstrated that the solubility of PIs was improved greatly by attaching bulky tert-butyl groups to the polyimide backbones, while their inherent thermal property was still kept [21–25]. Thus, highly soluble, transparent and thermo-stable PIs would be obtained if we introduce tert-butyl groups into aromatic ether diamine with four benzene rings. In this paper, a novel diamine containing tert-butyl groups and multiphenoxy units, 4,4′-(3-(tert-butyl)-4-anminophenoxy)diphenyl ether, was synthesized and polymerized with several commercial aromatic dianhydrides to produce a series of PIs via one-step polycondensation. The effects of tert-butyl groups and multiple phenoxy units on the physical-chemical properties of PIs were investigated and discussed in detail.
Experimental
Materials
2-tert-butylaniline (Wuxi TPW Pharmaceutical Technology Co. Ltd.) and m-Cresol (Sinopharm Chemical Reagent Co. Ltd.) was purified by the vacuum distillation before use. Pyromellitic dianhydride (PMDA, Sinopharm Chemical Reagent Co. Ltd.), 3,3′,4,4′-diphenylether tetracarboxylic dianhydride (OPDA, TCI Chemicals), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA, J&K Chemical Co. Ltd.) and 2,2-bis(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropane dianhydride (6FDA, J&K Chemical Co. Ltd.) were purified by recrystallization from acetic anhydride before use. Isoquinoline, triethylamine, 4,4′-oxydiphenol, I2, o-phenanthroline, CuI, K3PO4 and other materials were purchased and used without further purification.
Measurement
The 1H and 13C NMR spectra were obtained on a 400 MHz Varian NMR spectrometer with DMSO-d 6 and CDCl3 as solvent. The mechanical property of the polyimide films was determined by a CMT-4104 (SANS, Shenzhen, China) tensile tester at a drawing speed of 1 mm/min. The FTIR spectra were recorded on a Perkin-Elmer Fourier transform infrared spectrometer. The X-ray diffraction measurement was carried out on a Rigaku D/max-2200/PC X-ray diffractometer with Cu/K-α radiation. The number-average molecular weights and polydispersity index of the resulting PIs were determined by a TOSOH HLC-8320 gel permeation chromatography (GPC) analyzer (polystyrene calibration). Solubility was determined qualitatively by placing 10 mg polymer into 1 mL solvent at room temperature. The transparency of the polyimide films were performed on a Perkin-Elmer Lambda 20 UV–vis spectrometer. Differential scanning calorimetry (DSC) curves were obtained on a PE Pyris-1 thermal analyzer under N2 atmosphere at a heating rate of 20 °C/min from 40 °C to 430 °C. Thermogravimetric analysis (TGA) was performed on a PE Pyris-7 thermal analyzer under nitrogen at a heating rate of 20 °C/min from 50 °C to 800 °C. The molecular weight of diamine 1 was determined by a waters Q-Tof Premier mass spectrometry. The rates of water absorption was determined by immersing the dry films (about 250 mg) in 25 °C water for 24 h and calculated from the differences of the weights.
Monomer synthesis
2-tert-Butyl-4-iodophenylamine (1)
1 was synthesized from 2-tert-butylaniline according to our previous report [23]. Yield: 93%; 1H NMR (400 MHz, CDCl3): 7.47 (d, J = 2 Hz, 1H; Ar H), 7.29 (dd, J 1 = 8, J 2 = 2 Hz, 1H; Ar H), 6.42 (d, J = 8 Hz, 1H; Ar H), 1.39 (s, 9H; CH3).
N-(2-tert-butyl-4-iodophenyl)acetamide (2)
2.75 g (10 mmol) compound 1, 20 mL CH2Cl2, 1.11 g (11 mmol) triethylamine were added into 100 mL single-necked flask under magnetic stirring and the mixture was cooled to 0 °C. A solution of acetyl chloride (11 mmol acetyl chloride in 5 mL CH2Cl2) was dropped into the mixture within 15 min. After the addition, the reaction was further conducted at the room temperature for 8 h. Then, 20 mL saturated NaHCO3 solution was added to the mixture to quench the reaction and the mixture were extracted with 20 mL CH2Cl2 to get the organic layer, which was dried with anhydrous Na2SO4. After the filtration, the dark oil was obtained by the rotating evaporation. The crude product was recrystallized from ethanol to afford 2.57 g compound 2. Yield: 81%; 1H NMR (400 MHz, DMSO-d 6 ): 9.22 (s, 1H; −NH-), 7.64 (s, 1H; Ar H), 7.55 (d, J = 8.0 Hz, 1H; Ar H), 6.83 (d, J = 8.2 Hz, 1H; Ar H), 2.02 (s, 3H; CH3), 1.34–1.23 (m, 9H; CH3).
4,4′-(3-(tert-butyl)-4-acetamidophenoxy) diphenyl ether (3)
3.49 g (11 mmol) compound 2, 1.01 g (5 mmol) 4,4′-oxydiphenol, 0.95 g (5 mmol) CuI, 1.80 g (10 mmol) o-phenanthroline, 6.37 g (30 mmol) K3PO4 and 50 mL DMF were added to the three-necked flask under magnetic stirring. After the air in the three-necked flask was replaced with nitrogen, the mixture was warmed up to 120 °C to react under a closed system for 36 h. The mixture was cooled to room temperature and 50 mL ethyl acetate was added into it. After filtration by Buchner funnel, the mixture was concentrated under reduced pressure to get the black oil. Then it was purified by column chromatography with silica gel with a petroleum ether/ethyl acetate (v/v = 3/1, 2/1, 1/1, in sequence) as the eluent to give 1.68 g compound 3. Yield: 58%; 1H NMR (400 MHz, DMSO-d 6 ): δ (ppm) 9.13 (s, 2H; −NH-), 7.06 (s, 8H; Ar H), 7.03 (d, J = 2.8Hz, 2H; Ar H), 6.99 (d, J = 8.6 Hz, 2H; Ar H), 6.74 (dd, J 1 = 8.5, J 2 = 2.8 Hz, 2H; Ar H), 2.02 (s, 6H; CH3), 1.29 (s, 18H; CH3).
4,4′-(3-(tert-butyl)-4-aminophenoxy)diphenyl ether (4)
5.80 g (10 mmol) compound 3, 25 mL concentrated hydrochloric acid and 25 mL ethanol was added to a 200 mL single-neck flask under magnetic stirring. The reaction was conducted under reflux for 12 h. After the reaction finished, it was cooled to room temperature and the right amount of saturated NaOH aqueous solution was added to it to neutralize (pH = 7–8). After extraction with 20 mL ethyl acetate for three times, the combined organic layer was concentrated under reduced pressure to get the black oil. Then the crude product was purified by column chromatography with silica gel for two times with a petroleum ether/ethyl acetate (v/v = 6/1, 5/1, 4/1, in sequence) as the eluent to give 3.92 g compound 4. Yield: 79%; 1H NMR (400 MHz, DMSO-d 6 ): δ (ppm) 6.92 (d, J = 9.1 Hz, 4H; Ar H), 6.85 (d, J = 9.2 Hz, 4H; Ar H), 6.79 (d, J = 2.1 Hz, 2H; Ar H), 6.67 (d, J = 8.5 Hz, 2H; Ar H), 6.61 (dd, J 1 = 8.5 Hz, J 2 = 2.1 Hz, 2H; Ar H), 4.65 (s, 4H; −NH2), 1.31 (s, 18H; CH3). 13C NMR (101 MHz, DMSO-d 6 ): 154.96 (Ar C), 152.38 (Ar C), 147.35 (Ar C), 143.13 (Ar C), 134.47 (Ar C), 120.21 (Ar C), 118.68 (Ar C), 118.61 (Ar C), 118.49 (Ar C), 118.28 (Ar C), 34.64 (C), 29.69 (C). HPLC-MS (ESI, m/z): [M + H]+ calcd for C32H37N2O3, 497.2726; found, 497.2798.
Polymer synthesis
A typical synthesis procedure was displayed as follows (e.g., 4-OPDA). 496.3 mg (1 mmol) 1, 10 mL m-cresol, 310.2 mg (1 mmol) OPDA and two drops of isoquinoline was added to a 50 mL three necked flask charged with a nitrogen inlet and a mechanical stirring under N2 atmosphere. The reaction mixture was heated to 80 °C under stirring and kept at this temperature for another 12 h. Then the reaction temperature was raised successively to 120 °C for 12 h, 150 °C for 12 h and 180 °C for 12 h. The mixture was cooled to room temperature and diluted by 8 mL CHCl3 under stirring. Then the resulting viscous mixture was poured into 300 mL methanol under stirring to afford a fiber-like precipitate. The precipitate was collected by filtration through Buchner funnel and dried under vacuum at 160 °C for 24 h. At last, the crude product was reprecipitated twice from DMF into methanol to give the polyimide 4-OPDA.
Yield is 717 mg (93%); IR (cm−1): ν = 2965 (CH2-H), 1779 (C=O), 1725 (C=O), 1610, 1489, 1373 (C-N), 1236, 1213, 1099, 943, 814. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.01 (d, J = 8.2 Hz, 2H; Ar H), 7.59 - 7.54 (m, 2H; Ar H), 7.50 (dd, J 1 = 8.2, J 2 = 1.9 Hz, 2H; Ar H), 7.28 (d, J = 2.7 Hz, 2H; Ar H), 7.07 (dd, J 1 = 9.1 Hz, J 2 = 2.3 Hz, 8H; Ar H), 6.93 (d, J = 8.6 Hz, 2H; Ar H), 6.83 (dd, J 1 = 8.6 Hz, J 2 = 2.7 Hz, 2H; Ar H), 1.30 (s, 18H; CH3).
4-PMDA
Yield: 95%; IR (cm−1): ν = 2966 (CH2-H), 1780 (C=O), 1730 (C=O), 1600, 1491, 1374 (C-N), 1231, 1106, 943, 807, 732. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.50 (d, J = 7.2 Hz, 2H; Ar H), 7.31 (d, J = 2.5 Hz, 2H; Ar H), 7.08 (d, J = 9.1 Hz, 8H; Ar H), 6.96–6.91 (m, 2H; Ar H), 6.84 (d, J = 8.9 Hz, 2H; Ar H), 1.30 (s, 18H; CH3).
4-6FDA
Yield: 92%; IR (cm−1): ν = 2968 (CH2-H), 1786 (C=O), 1729 (C=O), 1609, 1493, 1372 (C-N), 1238, 1104, 944, 846, 725. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.04 (dd, J 1 = 8.0 Hz, J 2 = 1.7 Hz, 2H; Ar H), 7.98 (s, 2H; Ar H), 7.89 (t, J = 7.0 Hz, 2H; Ar H), 7.29 (d, J = 2.5 Hz, 2H; Ar H), 7.12 - 7.03 (m, 8H; Ar H), 6.93 (dd, J 1 = 8.6 Hz, J 2 = 3.1 Hz, 2H; Ar H), 6.83 (dd, J 1 = 8.6 Hz, J 2 = 2.6 Hz, 2H; Ar H), 1.31 (s, 18H; CH3).
4-BPDA
Yield: 97%; IR (cm−1): ν = 2966 (CH2-H), 1777 (C=O), 1723 (C=O), 1600, 1491, 1370 (C-N), 1232, 1099, 943, 843, 745. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.25 (s, 2H; Ar H), 8.13 - 8.07 (m, 4H; Ar H), 7.31 (d, J = 2.6 Hz, 2H; Ar H), 7.08 (dd, J 1 = 19.2 Hz, J 2 = 9.1 Hz, 8H; Ar H) 6.96 (d, J = 8.6 Hz, 2H; Ar H), 6.84 (dd, J 1 = 8.6 Hz, J 2 = 2.6 Hz, 2H; Ar H), 1.32 (s, 18H; CH3).
Membrane preparation
PI membranes with the different thickness were prepared as follows: PI solution (0.8 wt.% in NMP) was cast onto a clean glass plate (3 × 3 cm) and then dried at 100 °C in the air oven until NMP was removed. After cooling to room temperature, the membranes were soaked in deionized water and peeled off from the substrates and further dried at 180 °C under vacuum for 20 h. The thickness of membranes was from 5 to 10 μm, which were used to FTIR and UV–vis spectra measurement. Similarly, PI solution (1 wt.%) was cast onto the clean glass plates (2 × 3 cm) to prepare the transparent membrane with thickness from 20 μm to 30 μm, which was used to XRD measurement. PI solution (5 wt.%) was cast onto a clean glass plates (6 × 6 cm) to prepare transparent membrane with thickness from 30 to 45 μm, which was used to measure the mechanical property and water absorption of PIs.
Results and discussion
Monomer synthesis
The synthetic route of the novel diamine 4, 4,4′-(3-(tert-butyl)-4-aminophenoxy)diphenyl ether, was outlined in Scheme 1.

Synthesis of diamine 4
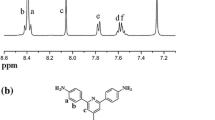
Firstly, 2-tert-butyl-4-iodophenylamine (1) was prepared from 2-tert-butylaniline by the iodination reaction. Then intermediate 1 was reacted with acetyl chloride to protect the amino group on the benzene ring to produce N-(2-tert-butyl-4-iodophenyl)acetamide (2). Next, in the presence of K3PO4 and DMF under the nitrogen atmosphere, the intermediate 3 was obtained from the intermediate 2 and 4,4′-oxydiphenol through Ullman coupling reaction with CuI as a catalyst and 1,10-phenanthroline as a ligand. This is the key step to significantly influence the yield of diamine 4. Thus the reaction time of Ullman coupling reaction was prolonged to 36 h to insure the high yield of intermediate 3. At last the diamine 4 was produced through the deprotection of acetyl in intermediate 3 and purified by recrystallization two times. The title diamine 4 was characterized by 1H NMR, 13C NMR, MS and DSC. The 1H and 13C NMR spectra of diamine 4 were presented in Fig. 1.

1H (a) and 13C (b) NMR spectra of diamine 4
As shown in Fig. 1a, the proton signals of amino group (Hg) appeared around 4.65 ppm, and five proton signals of benzene ring (Hb, Hc, Hd, He, Hf) were found from 6.61 to 6.92 ppm, whereas the proton signals of tert-butyl were observed at 1.31 ppm. Also, twelve carbon signals were found in the range from 29 to 155 ppm in Fig. 1b, which was in agreement with all carbon atoms in diamine 4. Furthermore, the molecular weight of diamine 4 was 496.2798 measured by MS, which was consistent with its theoretical value. The melting point of diamine 4 was around 110 °C, which was obtained from DSC analysis. All above results confirmed the chemical structure of diamine 4.
Polymer synthesis and characterization
Generally, polyimides can be synthesized from a diamine and a dianhydride through two-step procedure or one-pot approach [1]. According to our previous studies, the polymerization activity of the diamine will dramatically decrease due to the steric hindrance when tert-butyl groups are introduced into the ortho-position of the amino group [21, 22]. Thus, the two-step approach is not suitable for preparing polyimides with high molecular weights from diamine 4 with various dianhydrides. Here all the polyimides were synthesized from diamine 4 and a few commercial dianhydrides through the one-pot method in solution at high temperature as outlined in Scheme 2.

Synthesis of the PIs
When diamine 4 polymerized with the aromatic dianhydrides PMDA, BPDA, OPDA and 6FDA in m-cresol through the one-pot method, the process was carried out smoothly at the solid content of 10 wt % and the soluble PIs with high molecular weights were produced with the high yields (>90%). The resulting PIs was characterized by the means of GPC, 1H NMR, FT-IR and EA.
As was shown in Table 1, the number-average molecular weights (M n ) of the PIs were in the range from 2.1 × 104 to 3.7 × 104 with the polydispersity indices (PDIs) from 2.25 to 2.74. Apparently, the aromatic PIs with high molecular weight and relatively narrow PDI were still obtained by a one-pot method at high temperature even though the relatively low reactivity of the hindered aromatic diamine 4.
Figure 2 displayed the 1H NMR spectra of four resulting PIs in CDCl3 and all the signals were labeled. The results agreed with the chemical structure of the resulting PIs. In addition, no obvious signals of –OH or –NH were observed in the 1H NMR spectra, which indicated the imidization process was complete in the polymerization.

1H NMR spectra of the resulting PIs
The FT-IR spectra of the resulting PIs were displayed in Fig. 3. For all PI samples, the characteristic C=O vibration bands of imide ring could be observed at the wave number of 1778–1787 cm−1 and 1725–1730 cm−1, whereas the -OH vibration band and the C=O vibration band of poly(amic acid) were not detected at the wave number of about 3000 cm−1 and 1650 cm−1, respectively. These evidences also confirmed the imidization of the resulting PIs catalyzed by isoquinoline was completed at the one-pot method. All of the other characteristic bands were observed at wave number of 2965–2967 cm−1 for C-H in tert-butyl, 1371–1375 cm−1 for C-N, 1015–1104 cm−1 and 724–752 cm−1 for imide-ring. In addition, the elemental analysis results were consistent with the theoretically calculated values.

FT-IR spectra of the resulting PIs
Thermal properties
The thermal behavior of the resulting PIs was studied by means of TGA and DSC measurements. The TGA curves of all PIs were shown in Fig. 4 and the temperature at 5% weight loss of these PIs was shown in Table 1. Obviously, all the PIs were stable up to 450 °C. The weight loss temperature of 5% was in the range of 525–529 °C at nitrogen atmosphere. The char yields of all PIs at 800 °C were higher than 43 wt%. Compared with the previous PIs containing tert-butyl and phenoxy units, the inherent thermal stability of the resulting PIs was not sacrificed by introducing bulky pendant groups and multiple phenoxy units into their backbones [26–28].

TGA curves of the resulting PIs
The DSC curves of all PIs were presented in Fig. 5 and the corresponding T g s were also summarized in Table 1. According to our previous studies, the incorporation of tert-butyl into the polyimide backbone could retain its high T g, but multiple phenoxy units decreased it T g. Combining the effects of these two different units, the T g s of the resulting PIs were still in the range of 264–300 °C, which depended on the stiffness and polarity of the corresponding dianhydride. For example, 4-PMDA had a highest T g of 300 °C while 4-OPDA had a lowest T g of 264 °C.

DSC curves of the resulting PIs
Solubility
Generally, the solubility of polyimides depends on their chain packing ability and intermolecular interactions. Here we investigated the effect of tert-butyl group and multiple phenoxy units on the solubility of the resulting PIs at 10 mg/mL (PI/solvent) and room temperature or 60 °C in various solvents. The results were summarized in Table 2. The solubility of the resulting PIs was obviously enhanced and all of them could be dissolved in conventional organic solvents, such as NMP, DMF, DMAc, THF and CHCl3 at room temperature, including 4-PMDA with the most rigid dianhydride. Especially, 4-6FDA exhibited the most excellent solubility and even dissolved in toluene at room temperature. The enhanced solubility can be attributed to tert-butyl groups reducing the interaction between polyimide chains and multiple phenoxy units improving the flexibility of main chains of PIs. These two factors synchronously helped to the solvent molecules permeating into PI molecular chains and the solubility was indeed improved distinctly.
Mechanical behaviors and Water Absorption Rates
The mechanical property of the resulting PI films with the thickness about 40 μm was measured by CMT-4104 tensile tester at room temperature and the results were summarized in Table 3. All the resulting PI films had the tensile strength from 50 to 57.3 MPa, the tensile modulus from 1.94 to 2.48 GPa and the elongation at break of 4.3 to 12.1%. The relatively low elongation at break was attributed to amorphous glassy states of these PIs, because such kinds of polymers are usually somewhat brittle at the temperature far below T g. Furthermore, the water absorption rates of the resulting PIs were determined below 1% (Table 3). This can be attributed to the strong hydrophobic properties of tert-butyl. These PI films may have potential applications in the electrical fields due to their low water absorption rates [29].
X-ray diffraction data
The morphology of the resulting PI films (film thickness about 30 μm) was examined by X-ray diffractometer with 2θ from 5 to 40° and the diffraction spectra were exhibited in Fig. 6. All of them showed amorphous patterns in their X-ray diffraction curves. This indicated that the incorporation of tert-butyl and multiple phenoxy units into PIs increased the distance of PI main chains, reduced the stacking density and also enhanced the solubility of PIs [30].

X-ray diffraction curves of the resulting PI films
Optical properties
The UV–vis spectra of the resulting PI films with similar thickness (about 10 μm) are shown in Fig. 7 and their optical properties were outlined in Table 4. All of the resulting PIs films showed an excellent transmission above 90% in the visible region (400–760 nm) and their cut-off wavelengths ranged from 296 to 352 nm. This can be attributed to the bulky tert-butyl groups, which increased the intermolecular distance, weakened the PIs chain packing and restrained CTC formation, thus improving the optical transparency of PIs films.

UV–vis spectra of the resulting PI films
Conclusions
A series of soluble PIs with high transparency were prepared by introducing the bulky pendant groups tert-butyl and flexible multiple phenoxy units into their backbone. Our results further confirmed that the incorporation of bulky side groups and multiple phenoxy units could enhance solubility and improve transparency of the resulting PIs. In addition, multiple phenoxy units didn’t deteriorate heavily the inherent thermal properties of PIs. The strong and tough PI films could be prepared by solution-casting, which are potential candidates for micro-electronic or optical applications.
References
MittalEd KL (ed) (1984) Polyimides: synthesis, characterization and application. Plenum, New York
Xiao Y, Low BT, Hosseini SS, Chung TS, Paul DR (2009) Prog Polym Sci 34:561–580
Zhang A, Li X, Nah C, Hwang K, Lee MH (2003) J Polym Sci A Polym Chem 41:22–29
Feger C, Khojasteh MM, Htoo MS (eds) (1993) Advances in polyimide science and technology technomic. Lancaster
Chen CJ, Yen HJ, Chen WC, Liou GS (2011) J Polym Sci A Polym Chem 49:3709–3718
Ye YS, Huang YJ, Cheng CC, Chang FC (2010) Chem Commun 46:7554–7556
Liaw DJ, Wang KL, Huang YC, Lee KR, Lai JY, Ha CS (2012) Prog Polym Sci 37:907–974
Chun IS, Kim SY (2000) Macromolecules 33:3190–3193
Rusanov AL, Shifrina ZB (1993) High Perform Polym 5:107–121
Harris FW, Sakaguchi Y, Shibata M, Cheng SZD (1997) High Perform Polym 9:251–261
Ioakim KS, John AM (1996) Macromolecules 29:5313–5319
Grubb TL, Ulery VL, Smith TJ, Tullos GL, Yagci H, Mathias LJ, Langsam M (1999) Polymer 40:4279–4288
Yang CP, Su YY, Chen YC (2006) Eur Polym J 42:721–732
Chung CL, Tzu TW, Hsiao SH (2006) J Polym Res 13:495–506
Huang XH, Huang W, Yan DY (2012) Acta Polym Sin 5:552–560
Abbasi F, Mehdipour-Ataei S, Khademinejad S (2015) Des Monomers Polym 18:89–798
Revathi R, Prabunathan P, Devaraju S, Alagar M (2015) High Perform Polym 27:247–253
Liaw DJ, Liaw BY, Yu CW (2001) Polymer 42:5175–5179
Huang XH, Huang W, Fu LC, Yan DY (2012) J Polym Res 19:1–9
Tamai S, Yamaguchi A, Ohta M (1996) Polymer 37:3683–3692
Huang W, Yan DY, Lu QH (2001) Macromol Rapid Commun 22:1481–1484
Huang W, Yan DY, Lu QH, Tao P (2002) J Polym Sci A Polym Chem 40:229–234
Yi L, Li CY, Huang W, Yan DY (2015) J Polym Sci A Polym Chem 54:976–984
Huang XH, Huang W, Zhou YF, Yan DY (2011) Chin J Polym Sci 29:506–512
Li CY, Yi L, Xu ST, Huang W, Yan DY (2016) Acta Polym Sin 7:938–945
Liaw DJ, Liaw BY (1996) Polym J 28:970–975
Liaw DJ, Liaw BY (1997) J Polym Sci A Polym Chem 35:1527–1534
Dine Hart RA, Wright WW (2003) Makromol Chem Rapid 143:189–206
Imamura S, Yoshimura R, Izawa T (1991) Electron Lett 27:1342–1343
Yi L, Li CY, Huang W, Yan DY (2014) J Polym Res 21:1–10
Acknowledgements
The authors gratefully acknowledge the financial supports provided by the National Basic Research Program of China (No. 2014CB643604), Shanghai Key Projects of Basic Research (No. 16JC1403600).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, C., Yi, L., Xu, S. et al. Synthesis and characterization of polyimides from 4,4′-(3-(tert-butyl)-4-aminophenoxy)diphenyl ether. J Polym Res 24, 7 (2017). https://doi.org/10.1007/s10965-016-1168-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-016-1168-1