
Abstract
The benzothiadiazole/hexylthiophene, benzothiadiazole/hexylthiophene/N–hexylphenothiazine, benzothiadiazole/N-hexylphenothiazine alternating π-conjugated copolymers were synthesized via Pd-catalyzed cross-coupling reaction. The main structural differences among the three copolymers are the type of donor moiety (hexylthiophene and/or hexylphenothiazine). The polymer structures and photophysical properties were characterized by 1H NMR, 13C NMR, GPC, TGA, DSC, UV–vis absorption spectroscopy, PL spectroscopy, CV, and XRD measurement. The work is aimed at exploring the structural factors that could control the photophysical properties of copolymers in order to help in the rational design of polymers having specific physical properties used in optoelectronic devices. XRD of all copolymers showed a d-spacing range of 4.04 ~ 3.91 Å, reflecting the π-π stacking and some degree of crystallinity in their structure. Their PL spectra showed red and near infrared light, which nominates them as potential red and near infrared light-emitting materials for PLEDs. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were employed in an attempt to supplement and explained the experimental measurement. The preliminary photovoltaic prediction of the studied copolymers was also reported.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In contemporary years, with increasing demand on renewable energy, π-conjugated polymers have received a major level of importance due to their distinguished optical and electronic properties [1,2,3,4,5,6,7,8,9,10,11]. Such materials can be used in a variety of advanced technological applications as photovoltaic light-emitting diodes, and electrochromic devices [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16] owing to their low cost, consistency, and controlled effective properties (electronic, optical, stability, and conductivity). Thus, the improvement of synthetic procedures for the simple synthesis and/or modification of such organic materials are an attractive issue in organic synthesis. Conventionally, many π-conjugated polymers were synthesized using a variety of transition metal-catalyzed cross-coupling reactions (e.g., Kumada, Suzuki, Negishi, and Stille) that allow the formation of carbnon–carbon bonds [17,18,19,20,21,22,23,24]. However, former preparation of bifunctional organometallic reagents as monomers is demanded for these synthetic methods [25,26,27,28,29,30,31,32,33,34,35,36,37].
Recently, OPVs of alternating 2,1,3-benzothiadiazole donor–acceptor (D–A) based copolymers showed impressive power conversion efficiencies values (PCEs) near to 15% [38, 39] due to the proper electronic characteristic and efficient intramolecular charge transfer (ICT) [40,41,42,43,44,45]. Copolymerization of benzothiadiazole with various suitable arylenes [46, 47] can be used as a means to tune the HOMO/LUMO levels in the resulting polymers. The HOMO and LUMO energy level of π-conjugated polymer is essential for modifying charge injection processes in the luminescent devices. On the other hand, the electron-rich nature of phenothiazine contributes to the efficient electron donor and hole-transporting materials in polymers and organic molecules for photoinduced charge separation, and it has also been proven as a superior electron donor for reductive quenching [48,49,50]. Phenothiazine derivatives are known to generate more stable radical cations due to presence of two electron donor atoms (thiophene and nitrogen), also these derivatives generate more stable radical cations than other donor species such as thiophene [51]. The electron-rich nature of phenothiazine contributes for the efficient electron donor and hole transporting materials in polymers and organic molecules for photo-induced charge separation and it has been also proven as a superior electron donor for reductive quenching [52]. They are also characterized by electrical and thermal stability, with outstanding optoelectronic characteristics in the device [52,53,54].
The low-energy triplet excited state in conjugated polymers poses a substantial barrier to next-generation optoelectronic device applications. One advantage of conjugated polymer semiconductors is their strong absorption and emission, due to the almost total overlap of π and π* orbitals. However, the localized and overlapping wave functions result in a sizeable triplet-stabilizing exchange energy ΔEST of ≈ 0.7 eV [55,56,57].
We have previously reported on the synthesis of a wide variety of π-conjugated organic and polymeric molecules for electronic applications [58,59,60,61,62,63]. Thus, in this study, hexylthiophene and/or N-hexylphenothiazine were used as the electron donor, the benzothiadiazole serves as a robust electron affinity as the electron acceptor, with hexylthiophene rich in electrons connected to each terminal to increase the sufficient π-conjugation length, thereby designing a basic molecular structure that gives the polymer a low band gap. The photophysical and electrochemical characteristics of the synthesized benzothiadiazole based copolymers will be discussed in detail based on their components of repeating units. The DFT calculations to calculate the geometric and electronic structures were also discussed in detail. There are some previously published research works on similar polymers which have been synthesized which appeared promising properties and applied in different OPVs applications. Their properties were also close to our synthesized properties [64,65,66,67].
Experimental
Materials
Unless otherwise noted, all manipulations and reactions involving air-sensitive reagents were performed under a dry nitrogen atmosphere. All reagents and solvents were obtained from commercial sources and they dried using standard procedures before use, whenever required. Phenothiazine (2), 1-bromohexane, 4,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzo[c][1,2,5]thiadiazole) (4), and 3-hexylthiophene-2-boronic acid pinacol ester (5) were imported from Sigma-Aldrich. All simple organic chemical reactions were monitored by thin layer chromatograpy (TLC) for ensuring the completion.
Instrumentation
1H and 13C NMR spectra were measured on a Varian spectrometer (400 MHz for 1H and 100 MHz for 13C) in CDCl3 at 25 °C with TMS as the internal standard and chemical shifts were recorded in ppm units. The coupling constants (J) are given in Hz. Flash column chromatography was performed with Merck silica gel 60 (particle size 230–400 mesh ASTM). Microwave assisted polymerizations were performed in a focused microwave synthesis systemCEM (Discover S-Class System). The gel permeation chromatographic (GPC) analysis was carried with a Shimadzu (LC-20A Prominence Series) instrument; coupled with an UV detector (Shimadzu Corp., SPD-10A). Combination of Shodex KF-801 (30 cm, exclusion limit: Mn = 1.5 × 103, polystyrene) KF-802 (30 cm, exclusion limit: Mn = 5.0 × 103, polystyrene) and KF-803L (30 cm, exclusion limit: Mn = 7.0 × 104, polystyrene) columns (linear calibration down to Mn = 100) were used for molecular weight analysis. Chloroform was used as a carrier solvent (flow rate: 1 mL/min, at 30 °C) and calibration curves were made with standard polystyrene samples. The UV–vis absorption spectra were obtained using JASCO double beam UV–Vis-NIR scanning spectrophotometer (UV-780) on the pure polymer samples while the fluorescence spectra in solution were recorded using JASCO FP-8300 scanning spectrofluorometer and the fluorescence spectra of thin films were recorded on Kimmon Koha IK Series He-Cd Laser (320 nm). The thermal degradation temperature was measured using thermogravimetric analysis (TGA-TA instrument Q-50) under nitrogen atmosphere. Differential scanning calorimetry (DSC) was performed on a TA instrument (DSC-TA instrument Q-20) under nitrogen atmosphere at a heating rate of 10 °C/min. XRD experiments were performed with a Bruker D8 advanced model diffractometer and with Cu-Kα radiation (λ = 1.542 Å) at a generator voltage of 40 kV and a current of 40 mA. The CV measurements were performed on B-class solar simulator: Potentiostate/Galvanostate (SP-150 OMA Company). The supporting electrolyte was tetrabutylammonium hexafluorophosphate (TBAPF6) in acetonitrile (0.1 M) at a scan rate of 50 mV/s. A three-electrode cell was used; A Pt wire and silver/silver chloride [Ag in 0.1 M KCl] were used as the counter and reference electrodes, respectively. The CV measurements were calibrated using the ferrocene value of (–4.39 eV) as the standard. The polymer films for electrochemical measurements were spin coated from a polymer solution on ITO glass slides (10 mg/mL).
Results and discussion
Synthesis of precursory monomers and copolymers
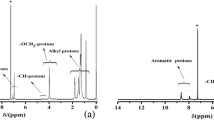
The precursory comonomer 4,7-bis(5-tributylstannyl-4,4'-hexylthiophene-2-yl)benzo[c][2,1,3]thiadiazole (1, Fig. 1) was readly prepared with in a chemical yield of 93% via our previously reported method [58] to be used subsequently as building block for synthesizing the target copolymers. 1H and 13C NMR spectra of comonomer 1 (Supporting Information) are fully consistent with the reported spectral data [58].

TGA thermograms of copolymers P1–P3

In another synthetic pathway, phenothiazine (2) was readily N-alkylated through its reaction with 1-bromohexane in N,N-dimethylformamide (DMF) as a solvent in the presence of potassium tert-butoxide as a base affording the desired product 10-hexyl-10H-phenothiazine (3, HPT) in 88% yield (Scheme 1).

Synthesis of N-hexylphenothiazine comonomers 4 and 7
Bromination of compound 3 with N-brmosuccinimde (NBS; 1/2 molar ratio) in dichloromethane afforded the corresponding 3,7-dibromo-N-hexylphenothiazine (4) in 87% yield. Moreover, Pd-catalyzed Suzuki cross-coupling reaction of 4 with 3-hexylthiophene-2-boronic acid pinacol ester (5) afforded the expected coupling product 10-hexyl-3,7-bis(3-hexylthiophene-2-yl)-10H-phenothiazine (6) in 88% yield. Brominating compound 6 with NBS (1/2 molar ratio) in DMF afforded the desired product 10-hexyl-3,7-bis(5-bromo-3-hexylthiophene-2-yl)-10H-phenothiazine (7) in 90% yield. The chemical structures of the synthesized organic compounds 3, 4, 6, and 7 were confirmed by elemental analysis as well as by 1H and 13C NMR spectroscopy and all data were found to be fully consistent with the proposed structures. Their spectral data are mentioned in the experimental part, and their corresponding spectral analyses are included in the supporting information.
The π-conjugated copolymers P1–P3 were readily synthesized as outlined in Scheme 2 via Pd-catalyzed cross-coupling reaction under microwave irradiation. Stille cross-coupling polymerization of equimolar amounts of comonomer processor 1 with 4,7-dibromobenzo[c]-1,2,5-thiadiazole 8 in DMF as a solvent in the presence of catalytic amounts of Pd(PPh3)4 afforded the corresponding copolymer P1 in 87% yield. However copolymer P1 has only benzothiadiazole (BT; acceptor) and hexylthiophene (HT; donor) units without additional donors. Pd-catalyzed Suzuki cross-coupling copolymerization of 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) (9) with comonomers 7 or 4 afforded the corresponding copolymers P2 and P3 in 88 and 92% yield, respectively (Scheme 2). Elemental and 1H NMR analyeses were used to prove the chemical structures of the obtained copolymers, and all data are entirely consistent with the proposed structures (see the experimental part for their spectral data and supporting information for their spectral analyses).

Synthetic routes of π–conjugated copolymers P1–P3. Polymerization conditions: (i) Stille cross-coupling: Pd(PPh3)4, DMF, microwave irradiation & (ii) Suzuki cross-coupling: Pd(PPh3)4, toluene, aq. K2CO3 (2 M), microwave irradiation
It is worth mention that copolymer P2 with the same order of donor and acceptor units in the main repeating units in polymer chains could also be synthesized by Stille cross-coupling copolymerization of comonomers 1 and 4 in dry DMF in the presence of Pd(PPh3)4 under microwave irradiation. Interstingly, the 1H NMR and UV–vis absorption spectral data of the resulting copolymer P2 that prepared through Suzuki and/or Stille cross-coupling conditions were found to be completely identical.
After precipitation into methanol, the crude copolymers were filtered off, washed extensively with methanol, followed by Soxhlet extraction with methyl alcohol and acetone successively to remove byproducts and oligomers. Gel permeation chromatography (GPC) was used to estimate the molecular weight and the molecular weight data are summarized in Table 1. Analysis of the copolymers P1–P3 showed that the palladium catalyst was removed entirely from the polymers.
All copolymers showed symmetrical unimodal SEC curves with relatively good weight- and number-average molecular weights (Mw and Mn, respectively) as well as molecular weight distributions (PDI) (Table 1). It is worth mention that the copolymer P2 obtained from the copolymerization of 1 and 4 had little lower Mw and Mn values (Table 1, in parentheses) than those observed for the same copolymer (P2) originating from copolymerization of 9 and 7 (Table 1). This was most likely due to the expected higher steric hindrance resulting from the outward hexyl side chains at the 4,4'-positions of the thiophene rings of comonomer 1 during the Stille cross-coupling. The observed molecular weights of the synthesized copolymer P1–P3 were found to be adequate for film processing. However, the film polymer formation was easily and readily fabricated from their solutions in most common organic solvents at room temperature. The excellent solubility of the synthesized copolymers (> 5 mg mL−1) could be attributed due to the presence of solubilizing hexyl chains either on thiophene and/or phenothiazine moieties.
Thermal properties
The thermal stability of copolymers P1–P3 was explored by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), under nitrogen atmosphere. TGA of copolymers reveals that the residual weights of copolymers P1–P3 are greater than 50% when the temperature was raised to 800 °C (Fig. 1 and Table 1).
The copolymers P1–P3 showed one-step decomposition process with the onset decomposition temperatures (Td, onset) at 385.0, 383.0, and 391.0 °C is corresponding to ~ 96.9, 95.9, and 97.4 weight % residues, respectively, (Table 1, Fig. 1) indicative of high thermal stabilities, which could be assigned to side-chain decomposition upon heating processes [68]. On the other hand, thermal decomposition maximum temperatures (Td,max; correspond to the maximum rate of weight loss) of copolymers P1–P3 were found to be located at 500.4, 533.6, and 511.0 °C, with remaining weights of ~ 82.03, 64.70, and 67.3%, respectively. Interestingly, TGA revealed that at the end process, the remaining weights of P1–P3 were found to represent ~ 68.8, 56.3, and 60.0% of the total weight of polymer samples, which we begin with it (it was represented by charing %).
The glass transition temperatures (Tg) of copolymers P1–P3 are also summarized in Table 1, while their DSC curves are presented in Fig. 2. The copolymer samples were heated up to 300 °C, and the DSC data were obtained from the second heating cycle. DSC analysis revealed that copolymers P1–P3 are amorphous materials with glass transition temperatures at 72.38, 112.27, 104.52 °C, respectively. The amorphous nature of the copolymers might be understood from the hexyl side chains on the thiophene and phenothiazine moieties protruding out of the polymer backbone planes.

DSC curves of copolymers P1–P3
Interestingly, the Tg of the copolymer P2 (incorporating BT, HT, and HPT moieties) was found to be higher than those of copolymers P1 and P3 (Table 1). This owes to the other intermolecular interactions and the increasing interchain regularity caused by introducing HT moiety between the BT and HPT moieties in the repeating units of the copolymer main chains. However, the high thermal stability of the copolymers could prevent the deformation of their morphology and the degradation of their polymeric active layer under applied electric fields. It is worth mention that the estimated Tg values were found to be above 50 ˚C, indicating that the three copolymers have good tolerance to the stages required in making devices whenever possible.
Optical properties
While the origin of the dual-band absorption sometimes encountered in D–A type semiconducting polymers remains a source of debate, two mainstream rationales were frequently proposed. A first assumption attributes the lower-energy optical transition to the presence of intermolecular charge-transfer excitons occurring on the presence of covalently bound D–A segments along the backbone. A second assumption considers the presence of low-lying unoccupied energy levels, strictly localized on the electron-deficient heterocycles, yet forming a discrete "band" of easily accessed energy states within the band gap of the conjugated system in its ground state. In both cases, the higher-energy transitions appear localized on the most electron-rich building units incorporated along the polymer backbone with a clear dependence on their relative concentration to the electron-deficient heterocycles. The photophysical characteristics of the copolymers P1–P3 were investigated by ultraviolet–visible (UV–vis) absorption and photoluminescence (PL) spectroscopy in diluted toluene solutions and as thin films prepared from their solutions on glass slides. The optoelectronic properties (UV–vis and PL spectral data) are summarized in Table 2.
The plots of the UV–vis absorptions for the copolymers P1–P3 (solutions and thin films) are depicted in Fig. 3. As expected, each copolymer exhibited a dual-band of absorption, which could be assigned to the π-π∗ transition of the conjugated backbone and ICT interactions between the donor and acceptor units [51]. The UV–vis analysis of copolymer solutions of P1–P3 (Fig. 3) exhibited λmax1 absorption bands at 321.1, 358.9, and 307.8 nm and λmax2 at 514.3, 499.1, and 467.5 nm, respectively (Table 2). Moreover, copolymer thin films (P1–P3) exhibited also two major absorption bands: λmax1 at 326.7, 368.1, and 344.5 nm and λmax2 at 532.7, 527.1, and 504.9 nm, respectively. Interestingly, both λmax1 and λmax2 in the film state are relatively red-shifted relative to those of the solution state. It is worth mention that the absorption bands characteristic for 2,1,3-benzothiadiazole adsorbs at ~ 306 nm [52] and for phenothiazine ring are localized at ~ 318 nm [57,58,59,60,61,62,63]. However, the extent of conjugation between neighboring BT units and either HT or HPT ones will largely depend on the twist angle. The twisting between all adjacent units of the tested molecules reduces the overlap of p orbitals on the twisting regions because these orbitals will not be completely parallel. Accordingly, this will reduce the extent to which the waves are delocalized across the molecular backbone and therefore a higher optical band gap will be obtained. As shown in the absorption spectra of P1, which has the lowest twist angle, it is clear that P1 has the reddest shifted λmax2 and λonset in solution and film.

UV–vis absorption spectra of polymers P1–P3 in solutions and films
As we notice that the polymer structures of P1–P3 are differing in the type of donor moiety (HT and/or HPT). The UV–vis absorption spectra of copolymers P1 and P2 show extended absorption in the solid state. This is properly due to their ability to form crystals and aggregations through the thin film leading to an increase in the intermolecular interactions between neighboring molecules in the film state, which leads to an increase in the conjugation due to the increased π-π stacking of the polymer backbone in the solid polymer state [69, 70] and consequently low optical band gaps (\({E}_{g}^{op}\)). The UV–vis absorption spectra of copolymers P1–P3 in the thin-film state showed two broad bands, and the shoulder results in cut-off wavelengths (absorption onsets; λonset) of 645.5, 629.1, and 610.7 nm, respectively, corresponding to optical band gaps of 1.92, 1.97, and 2.03 eV. However, the absorption onset wavelengths of copolymer solutions of P1–P3 were 597.3, 582.7, and 555.4 nm, which correspond to the optical band gaps of 2.08, 2.13, and 2.23 eV, respectively (Table 2).
It is worth mention that the onset absorptions (λonset) of the copolymer thin films are also relatively red-shifted compared to their solutions (Table 2). For example, the red-shift of onset absorption was about 48.2 nm for P1, 46.4 nm for P2, and 55.3 nm for P3 when compared to their corresponding values in solutions (Table 2). The red-shifts of absorption maxima and onset absorptions could be attributed to the molecular aggregation in solid-state. This is attributed to the larger red-shift of onset absorption than absorption maxima. It is worth mention that copolymer P3 showed a significant large extended UV–vis absorption (in both solution and thin film) when compared to previously reported N-alkylphenothiazine-benzothiadiazole co-oligomer [65].
The fluorescence emission spectra of copolymers P1–P3 acquired by irradiative excitation at their respective wavelengths of the absorption maxima in the toluene solution at a concentration of 10–6 M and their spin-coated films on glass slides are given in Fig. 4. The fluorescence emission peak maxima (PL max) of copolymer solution and film are also shown in Table 2. Upon photoexcitation of copolymers P1–P3 (1 \({\mu }{M}\)) in the toluene solution, they exhibited characteristic intense emission maxima peaks located at 630.9, 635.1 and 638.5 nm, respectively. The emission maxima peaks of spin coated polymer films of (P1 − P3) are red-shifted by 57.9 nm, 54.7 nm, and 51.9 nm, respectively, when compared to their emission maxima peaks in solutions, indicating an increase of the conjugation length upon chain desolvation. This evidence may be attributed to an appreciable degree of self-organization experienced by the polymer in the solid-state. The PL spectra for the polymer solutions and the corresponding spin coated films, while being similar in shape to each other, show evident structuration and differ markedly from the absorption ones.

Photoluminescence spectra of copolymers P1–P3 in solution (a) and spin coated films (b)
Electrochemical properties
Cyclic voltammetry (CV) was employed to examine the electrochemical properties and determine the frontier molecular orbital energies, EHOMO and ELUMO, of the synthesized copolymers. The CV curves of copolymers P1 − P3 are presented in Fig. 5, and the CV revealed data are listed in Table 3. The EHOMO and ELUMO of the copolymers were determined from their cast films on ITO glass substrates. They were calculated according to the empirical formulas: [71, 72] EHOMO = –(Eox + 4.39) eV, and ELUMO = –(Ere + 4.39) eV; where Eox and Ere are the onset oxidation and reduction potentials of the polymers, respectively, vs. SCE (Ag/AgCl).

Cyclic voltammograms of copolymers P1 − P3 thin films recorded in 0.1 M Bu4NPF6/acetonitrile (supporting electrolyte) at a scan rate of 50 mV/s
As shown in Table 3, the estimated electrochemical band gaps (\({E}_{g}^{ec}\)) for copolymers P1 − P3 are very close to each other (1.96 ~ 2.08 eV). Interestingly, there is a slight difference in the HOMO or LUMO energy levels by incorporating the N-hexylphenothiazine instead of hexylthiophene donor unit in the polymer chains (Table 3). As the EHOMO of both copolymers were found to be below the air oxidation threshold (ca. –5.27 eV), [73] the two polymers may show good stabilities toward air and oxygen (a prerequisite when considering device application). It was observed that the LUMO energy levels of copolymers P1 − P3 (−3.41, −3.30, and −3.23 eV, respectively) are higher than those of PC61BM (≈ –3.91 eV), which indicates the efficient photoinduced electron transfer from the polymers (as a donor) to PCBM (as acceptor) is allowed [74].
Based on the CV results, the synthesized copolymers showed promising electrochemical properties as polymer donor materials. Interestingly, although the estimated \({E}_{g}^{ec}\) of polymers, occasionally, were reported to be somewhat higher than those corresponding values of \({E}_{g}^{op}\) (originated from the interface energy barriers present between the polymer films and the electrode surfaces), [75,76,77] the estimated \({E}_{g}^{ec}\) and \({E}_{g}^{op}\) of all copolymers were found be very close to each other indicating that the polymer thin films were spin-coated perfectly.
X-ray diffraction (XRD) study
To study the crystallinity of the synthesized copolymers, XRD was performed, and Fig. 6 shows the XRD patterns of powders of copolymers P1 − P3. The d-spacing was obtained according to Bragg's equation, nλ = 2dhkl sinθ, where λ is the radiation wavelength, dhkl is the specific lattice spacing, and θ is the diffraction angle. All copolymers (P1 − P3) show a first broad peak at ~ 22.20°, 22.70°, and 21.94° corresponding to a d-spacing of 3.99, 3.91, and 4.04 Å, respectively. This is believed due to the π-π stacking of the polymer backbone [78]. Since copolymers P1 and P2 contain 3-hexylthiophene in their polymer backbone structures, the π-π stacking distances of these polymers were similar to P3HT (d2 = 3.8 Å). Moreover, copolymers P1 − P3 showed second but also sharp peaks at 35.30°, 34.95°, and 39.30° corresponding to a d-spacing of 2.54, 2.56, and 2.29 Å, respectively, indicating the presence of some crystallinity. There is no apparent peak in a small angel region, for copolymers P1 and P3, while copolymer P2 shows a sharp peak at around 11.25°, by which it reveals that the distance between polymer main chains separated by alkyl side chains is 7.80 Å [79].

X–ray diffraction patterns of the copolymers P1–P3 in solid films
Overall, the low diffraction intensity of the π-stacking peaks in combination with their broad peaks (between 15° to 30°) suggests that these polymers have rather low crystallinity [80]. However, the presence of π-π stacking distance at the wide-angle region is related to flexible side chains and electrostatic interaction between D and A moieties [81].
A computational study
The density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations using B3LYP/6-31G(d,p) level [82,83,84,85] model chemistry were performed using Gaussian 16 code [86]. The influence of the replacement of benzothiadiazole by N-alkylphenothiazine on the geometries and electronic properties of P1, was performed. Moreover, we explored the effect of structural change from P2 to P3. In this context, long hexyl side chains have been simplified to methyl to construct molecular models that are computable with great precision. Figure 7 shows the optimized molecular geometries of the three comonomron. The calculations found that the interconnection between each of two adjacent subunits lies in the range of 1.45–1.48 Å, revealing that these links have a double bond characteristic. This might be a plausible marker of ICTs within their molecular backbone. All optimized structures adopt a more nonplanar conformation. The position of the alkyl group of thiophene moiety affects directly on the twist angle.

B3LYP-optimized geometries of the investigated comonomers. The dihedral angles among the moieties were assigned. The atom symbols are provided below the figure
The electron density topology and FMO energies of a conjugated polymer are the playmaker in most photophysical processes such as intramolecular / intermolecular charge transfer, light absorption / emission and charge / extraction / trapping injection as well as electrochemistry [87]. The FMO topology of the investigated compounds are presented in Fig. 8. The HOMO and LUMO wave functions of P1 are delocalized mainly over its entire π-conjugated backbone. The HOMO has an anti-binding character between consecutive subunits, whereas the LUMO indicates a binding character between the subunits. Thus, the lowest-lying singlet states are corresponding to the electronic transition of π–π* type. On the other hand, the HOMO wave function of P2 and P3 is localized on the donor moiety, whereas in contrast, the LUMO wave function is predominantly localized on the electron-deficient benzothiadiazole. This can be considered as another visual mark for ICT character. Although it is hard to relate the dipole properties to the photovoltaic performance of OPV materials, some studies suggest large dipoles moments are beneficial for charge separation in D–A blends [88] and achieving fill factors (FFs) in the devices [89]. The calculations showed that the molecular dipole moment has this order: P1 < P2 < P3. On the other hand, the computed orbital energies of HOMO and LUMO are overestimated by 0.48 and 0.91 eV, respectively, leading eventually to an underestimated energy gap by 0.44 eV than the corresponding cyclic voltammetry results, see Tables 3 and 4.

The FMO topology of the studied comonomers
The Bulk heterojunction organic solar cells (BHJ) depends on the intermolecular charge transfer in a blend made from donor organic materials such as conjugated polymers and acceptor materials such as fullerene. [6, 6]-Phenyl-C61-butyric acid methyl ester (PCBM(60)) is one of the most broadly used as an acceptor in solar cell devices [79]. A preliminary prediction of the photovoltaic properties of the investigated compounds as donor blended with PCBM(60) can be seen from the power of injection of photoelectron from the LUMO of polymer to the LUMO of PCBM(60), Fig. 9. The difference in the LUMO energy levels of the studied compounds (P1-P3) and PCBM(60), ΔELUMOs, was in the range of 1.02 to 1.38 eV, suggesting that the photoexcited electron transfer from the studied molecules to the acceptor PCBM(60) may be sufficiently efficient to be useful in photovoltaic devices [90] especially the cell that containing P1. On the other hand, the power conversion efficiency (PCE) can be calculated according to the following equation [91]:

The absolute energy of the FMO of comonomers and PCBM(60)
where Pinc is the incident power density, Jsc is the short-circuit current, Voc is the open-circuit voltage, and FF denotes the fill factor. The maximum open-circuit voltage (Voc) of the BHJ solar cell is related to the difference between the HOMO of the electron-donating polymer and the LUMO of the electron-accepting fullerene, taking into account the energy lost during the photo-charge generation [92, 93]. The theoretical values of open-circuit voltage Voc have been calculated from the following expression:
The calculated Voc of P1-P3 ranges from 0.83 eV to 1.19 eV. These values are higher than 0.3 eV, which guarantees efficient exciton split and charge dissociation at the donor/acceptor interface [3] and suggest the polymers under study are good candidates for photovoltaic application.
On the other hand, the short-circuit current density (Jsc) is another critical parameter for evaluating the BHJ solar cell performance as it is directly related to the power conversion efficiency. It can be identified with the following expression [94]:
where LHE(λ) is the light collection efficiency at a specific wavelength, \(\phi\) inj is the electron injection efficiency, and ηcoll is the charge collection efficiency. The latter is considered as constant. Generally, a high LHE is needed to get maximum photocurrent [95]. Knowing that f is the oscillator strength corresponding to the maximum absorption wavelength (λmax), the parameter (LHE) can be determined as follows [96]:
The calculated values of LHE are listed in Table 4. These values are in a small range (0.308–0.838). They decrease in the following order 0.838 (P1) > 0.712 (P2) > 0.308 (P3). It indicates that replacing the strong acceptor unit, such as benzothiadiazole, with a weaker one, such as phenothiazine, is not useful to enhance the photocurrent response. The calculated Voc of P1–P3 ranges from 0.83 eV to 1.19 eV showing the influence of structural changes. We noted that P1 has the best values of Voc among the studied compounds because it has the lowest optical band gap. These obtained values are sufficient for possible efficient electron injection which guarantees efficient exciton split and charge dissociation at the donor/acceptor interface [3] and suggest the polymers under study are good candidates for photovoltaic application.” However, the ϕinj and ηcoll also correlate with Jsc. Actually, Jsc composes from three components, LHE(λ), ϕinj and ηcoll. The ηcoll is the charge collection efficiency which is considered constant for the three OPV cells. Because LHE(λ) is the most dominant components of the short circuit current (Jsc), we only calculated LHE(λ) to discriminate among the studied cells.
TD-DFT calculations were performed to gain insights into the excited state properties of the comonomers. The calculated excited-state vertical transition energies by nanometer, oscillator strengths, and transition electronic configurations are given in Table 4. It can be seen that the calculations reproduce experimental absorption peaks. All the comonomers show mainly two optical transitions, one in the range of 517–534 nm, arising from ICT in the D–A segment, and the other second peak occurs in the range between 316–376 nm as a result of delocalized π → π* transition. After examining the predominant component of the molecular orbitals involved in the pertinent transitions, one can see that the lowest singlet S0 → S1 excited state with high-oscillator strength (between 0.93–1.86) corresponds predominantly to HOMO → LUMO transition (90–98%). At the same time, the other transition corresponds mainly to the HOMO → LUMO + 2 transition (82–90%).
Conclusions
By applying the donor–acceptor alternating strategy, three benzothiadiazole based copolymers (P1–P3) were synthetized. Benzothiadiazole serve as acceptor unit whereas, hexylthiophene and/or N-hexylphenothiazine act as donor units. The copolymers showed good thermal stability with residual weights greater than 50% upon raising the temperature to 800 oC. A broad absorption band was observed in the visible region with optical band gaps ranging from 1.92 ~ 2.03 eV in thin films. Cyclic voltammetry measurements showed that these copolymers are good as electron donor materials with electronic band gaps of 2.0 ~ 2.08 eV. Moreover, their PL spectra showed red and near infrared light, which nominates them as potential red and near infrared light-emitting materials for PLEDs. XRD pattern of copolymer P2 showed a sharp peak at around 2θ = 11.25°, which may be attributed to increased alkyl chains in structure when compared to P1 and P2. The signs of ICT could be seen from inter-distances in the range of 1.45 Å ~ 1.48 Å between every two adjacent subunits and the FMO topology. The DFT findings suggested that the photoexcited electron transfer from the studied polymers to the acceptor PCBM(60) may be sufficiently efficient to be useful in photovoltaic devices. Overall, in view of their optoelectronic properties, the synthesized copolymers can be coincided with particular interest in view of possible practical applications as optically emitting materials and organic photovoltaic devices. Further work in applications of these polymers in the luminescent devices and laser applications is in progress.
References
Bundgaard E, Krebs FC (2007) Sol Energy Mater Sol Cells 91(11):954–985
Günes S, Neugebauer H, Sariciftci NS (2007) Chem Rev 107(4):1324–1338
Thompson BC, Fréchet JM (2008) Angew Chem Int Ed 47(1):58–77
Scherf U, Dieter N (2008) Adv Polym Sci. Springer, Berlin
Huo L, Hou J (2011) Polym Chem 2(11):2453–2461
Kroon R, Lenes M, Hummelen JC, Blom PW, De Boer B (2008) Polym Rev 48(3):531–582
Chen J, Cao Y (2009) Acc Chem Res 42(11):1709–1718
Cheng YJ, Yang SH, Hsu CS (2009) Chem Rev 109(11):5868–5923
Boudreault PLT, Najari A, Leclerc M (2011) Chem Mater 23(3):456–469
Zhou H, Yang L, You W (2012) Macromolecules 45(2):607–632
Li Y (2012) Acc Chem Res 45(5):723–733
Dhanabalan A, van Duren JK, van Hal PA, van Dongen JL, Janssen RAJ (2001) Adv Func Mater 11(4):255–262
Aldakov D, Palacios MA, Anzenbacher P (2005) Chem Mater 17(21):5238–5241
Kono T, Kumaki D, Nishida JI, Sakanoue T, Kakita M, Tada H, Yamashita Y (2007) Chem Mater 19(6):1218–1220
Nielsen CB, Angerhofer A, Abboud KA, Reynolds JR (2008) J Am Chem Soc 130(30):9734–9746
Beaujuge PM, Ellinger S, Reynolds JR (2008) Nat Mater 7(10):795–799
Stille JK (1986) Angew Chem, Int Ed Engl 25(6):508–524
Miyaura N, Suzuki A (1995) Chem Rev 95(7):2457–2483
Littke AF, Fu GC (2002) Angew Chem Int Ed 41(22):4176–4211
Meijere AD, Diederich F (2004). Metal-catalyzed cross-coupling reactions. https://doi.org/10.1002/9783527619535
Carsten B, He F, Son HJ, Xu T, Yu L (2011) Chem Rev 111(3):1493–1528
Chen TA, Wu X, Rieke RD (1995) J Am Chem Soc 117(1):233–244
Wang X, Perzon E, Delgado JL, de la Cruz P, Zhang F, Langa F, Inganäs O (2004) Appl Phys Lett 85(21):5081–5083
Wu PT, Xin H, Kim FS, Ren G, Jenekhe SA (2009) Macromolecules 42(22):8817–8826
Godula K, Sames D (2006) Science 312(5770):67–72
Alberico D, Scott ME, Lautens M (2007) Chem Rev 107(1):174–238
Chen X, Engle KM, Wang DH, Yu JQ (2009) Angew Chem Int Ed 48(28):5094–5115
Ackermann L, Vicente R, Kapdi AR (2009) Angew Chem Int Ed 48(52):9792–9826
Roger J, Gottumukkala AL, Doucet H (2010) ChemCatChem 2(1):20–40
Borghese A, Geldhof G, Antoine L (2006) Tetrahedron Lett 47(52):9249–9252
Mohanakrishnan AK, Amaladass P, Clement JA (2007) Tetrahedron Lett 48(4):539–544
Amaladass P, Clement JA, Mohanakrishnan AK (2007) Tetrahedron 63(41):10363–10371
Mori A, Sugie A (2008) Bull Chem Soc Jpn 81(5):548–561
Bellina F, Rossi R (2009) Tetrahedron 65(50):10269
Liu CY, Zhao H, Yu HH (2011) Org Lett 13(15):4068–4071
Baghbanzadeh M, Pilger C, Kappe CO (2011) J Org Chem 76(19):8138–8142
Takita R, Fujita D, Ozawa F (2011) Synlett 2011(07):959–963
Yuan J, Zhang Y, Zhou L, Zhang G, Yip HL, Lau TK, Zou Y (2019) Joule 3(4):1140–1151
Ma L, Xu Y, Zu Y, Liao Q, Xu B, An C, Hou J (2020) SCIENCE CHINA Chem 63(1):21–27
Beaujuge PM, Amb CM, Reynolds JR (2010) Acc Chem Res 43(11):1396–1407
Scharber MC, Mühlbacher D, Koppe M, Denk P, Waldauf C, Heeger AJ, Brabec CJ (2006) Adv Mater 18(6):789–794
Peng Q, Liu X, Su D, Fu G, Xu J, Dai L (2011) Adv Mater 23(39):4554–4558
You J, Dou L, Yoshimura K, Kato T, Ohya K, Moriarty T, Yang Y (2013) Nat Commun 4(1):1–10
Zhao X, Lv H, Yang D, Li Z, Chen Z, Yang X (2016) J Polym Sci A Polym Chem 54(1):44–48
Li Z, Zhang T, Xin Y, Zhao X, Yang D, Wu F, Yang X (2016) J Mater Chem A 4(47):18598–18606
Herguth P, Jiang X, Liu MS, Jen AKY (2002) Macromolecules 35(16):6094–6100
Witker D, Reynolds JR (2005) Macromolecules 38(18):7636–7644
Jenekhe SA, Lu L, Alam MM (2001) Macromolecules 34(21):7315–7324
Bates WD, Chen P, Dattelbaum DM, Jones WE, Meyer TJ (1999) J Phys Chem A 103(27):5227–5231
Pfennig BW, Chen P, Meyer TJ (1996) Inorg Chem 35(10):2898–2901
Yun DH, Yoo HS, Seong KH, Lim JH, Park YS, Wo JW (2014) Appl Chem Eng 25(5):487–496
Padhy H, Huang JH, Sahu D, Patra D, Kekuda D, Chu CW, Lin HC (2010) J Polym Sci A Polym Chem 48(21):4823–4834
Choi JY, Kim DH, Lee B, Kim JH (2009) Bull Kor Chem Soc 30(9):1933–1938
Son SK, Choi YS, Lee WH, Hong Y, Kim JR, Shin WS, Kang IN (2010) Polym Chem 48(3):635–646
Köhler A, Beljonne D (2004) Adv Func Mater 14(1):11–18
Monkman AP, Burrows HD, Hartwell LJ, Horsburgh LE, Hamblett I, Navaratnam S (2001) Phys Rev Lett 86(7):1358
Andernach R, Utzat H, Dimitrov SD, McCulloch I, Heeney M, Durrant JR, Bronstein H (2015) J Am Chem Soc 137(32):10383–10390
El-Shehawy AA, Abdo NI, El-Barbary AA, Lee JS (2011) Eur J Org Chem 2011(25):4841–4852. https://doi.org/10.1002/ejoc.201100182
Abdo NI, El-Shehawy AA, El-Barbary AA, Lee JS (2012) Eur J Org Chem 2012(28):5540–5551. https://doi.org/10.1002/ejoc.201200769
Abdo NI, Ku J, El-Shehawy AA, Shim HS, Min JK, El-Barbary AA, Lee JS (2013) J Mater Chem A 1(35):10306–10317. https://doi.org/10.1039/C3TA11433C
El-Shehawy AA, Abdo NI, El-Barbary AA, Choi JW, El-Sheshtawy HS (2018) J Mater Sci Nanomater 2(103):2
El‐Shehawy AA, Abdo NI, El‐Hendawy MM, Abdallah ARI, Lee JS (2020) J Phys Org Chem e4063. https://doi.org/10.1002/poc.4063
El‐Shehawy AA, Abdu ME, El‐Hendawy MM, El‐Khouly M, Sherif MH, Moustafa HY (2020) J Phys Org Chem e4158. https://doi.org/10.1002/poc.4158
Liu CH, Chen SH, Chen Y (2006) J Polym Sci A Polym Chem 44(12):3882–3895
Nowakowska-Oleksy A, Cabaj J, Olech K, Sołoducho J, Roszak S (2011) J Fluoresc 21(4):1625–1633
Doğanci E, Gorur M. Journal of the Turkish Chemical Society Section A: Chemistry 3(3):565–582. https://doi.org/10.18596/jotcsa.28202
Li Y, Xue L, Li H, Li Z, Xu B, Wen S, Tian W (2009) Macromolecules 42(13):4491–4499
Anant P, Lucas NT, Jacob J (2008) Org Lett 10(24):5533–5536. https://doi.org/10.1021/ol8022837
Agrawal S, Pastore M, Marotta G, Reddy MA, Chandrasekharam M, Angelis F (2013) J Phys Chem C 117:9613–9622
Son SK, Choi YS, Lee WH, Hong YT, Kim JR, Shin WS, Moon SJ, Hwang DH, Kang IN (2010) J Polym Sci A Polym Chem 48:635–646
De Leeuw DM, Simenon MMJ, Brown AR, Einerhand REF (1997) Synth Met 87(1):53–59
Usta H, Risko C, Wang Z, Huang H, Deliomeroglu MK, Zhukhovitskiy A, Marks TJ (2009) J Am Chem Soc 131(15):5586–5608
McNeill CR, Halls JJ, Wilson R, Whiting GL, Berkebile S, Ramsey MG, Greenham NC (2008) Adv Func Mater 18(16):2309–2321
Li Z, Feng K, Liu J, Mei J, Li Y, Peng Q (2016) J Mater Chem A 4(19):7372–7381
Johansson T, Mammo W, Svensson M, Andersson MR, Inganäs O (2003) J Mater Chem 13(6):1316–1323
Hou J, Tan ZA, Yan Y, He Y, Yang C, Li Y (2006) J Am Chem Soc 128(14):4911–4916
Shang H, Fan H, Shi Q, Li S, Li Y, Zhan X (2010) Sol Energy Mater Sol Cells 94(3):457–464
Tong J, Zhang L, Li F, Wang K, Han L, Cao S (2015) RSC Adv 5(107):88149–88153
Blouin N, Michaud A, Gendron D, Wakim S, Blair E, Neagu-Plesu R, Leclerc M (2008) J Am Chem Soc 130(2):732–742
Ding P, Zou Y, Chu CC, Xiao D, Hsu CS (2012) J Appl Polym Sci 125(5):3936–3945
Lu W, Kuwabara J, Kuramochi M, Kanbara T (2015) J Polym Sci A Polym Chem 53(11):1396–1402
Becke AD (1993) J Chem Phys 7(98):5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37(2):785
Vosko SH, Wilk L, Nusair M (1980) Can J Phys 58(8):1200–1211
Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) J Phys Chem 98(45):11623–11627
Gaussian 16, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al (2016) Gaussian, Inc., Wallingford CT
Bronstein H, Nielsen CB, Schroeder BC et al (2020) Nat Rev Chem 4:66–77. https://doi.org/10.1038/s41570-019-0152-9
Carsten B, Szarko JM, Son HJ, Wang W, Lu L, He F, Yu L (2011) J Am Chem Soc 133(50):20468–20475
Gao W, Zhang M, Liu T, Ming R, An Q, Wu K, Zhang F (2018) Adv Mater 30(26):1800052
Gadisa A, Svensson M, Andersson MR, Inganäs O (2004) Appl Phys Lett 84(9):1609–1611
Morvillo P (2009) Sol Energy Mater Sol Cells 93(10):1827–1832
Narayan MR (2012) Renew Sustain Energy Rev 16(1):208–215
Dou L, Liu Y, Hong Z, Li G, Yang Y (2015) Chem Rev 115(23):12633–12665. https://doi.org/10.1021/acs.chemrev.5b00165
El Assyry A, Jdaa R, Benali B, Addou M, Zarrouk A (2015) J Mater Environ Sci 6(9):2612–2623
Youssef AA, Bouzzine SM, Fahim ZME, Sıdır İ, Hamidi M, Bouachrine M (2019) Phys B 560:111–125. https://doi.org/10.1016/j.physb.2019.02.004
Zhang ZL, Zou LY, Ren AM, Liu YF, Feng JK, Sun CC (2013) Dyes Pigm 96(2):349–363. https://doi.org/10.1016/j.dyepig.2012.08.020
Acknowledgements
This work was financially supported by the Science & Technology Development Fund (STDF–Egypt) through the STDF-NRG Project (Project ID: 7973). It was also financially supported by the Academy of Science and Research & Technology (ASRT), Egypt, Grant no 6371 under the project Science UP. Prof. Ashraf A. El-Shehawy would like also to acknowledge the generous support from Kafrelsheikh University Research Program (ID: KFSU-3-13-04).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
El-Shehawy, A.A., El-Hendawy, M.M., Attia, A.M. et al. Synthesis, photophysical properties, and computational studies of benzothiadiazole and/or phenothiazine based donor/acceptor π-conjugated copolymers. J Polym Res 28, 268 (2021). https://doi.org/10.1007/s10965-021-02621-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-021-02621-y