
Abstract
In this article, starch phosphate-g-poly (methacrylic acids) (SP-g-PMAAs) were synthesized by dual modification. Firstly starch phosphate (SP) was synthesized by phosphorylation of native starch (NS) with mono sodium-phosphate and di sodium-phosphate. Afterwards, methacrylic acid (MAA) was grafted onto SP via free radical polymerization to synthesize SP-g-PMAAs. The structure of the SP and SP-g-PMAAs was investigated by phosphorus nuclear magnetic resonance, proton nuclear magnetic resonance and fourier-transform infrared spectroscopy. The X-ray diffraction studies showed that the modification diminished the crystalline nature of SP and SP-g-PMAAs. The SEM images showed spherical or polyhedral smooth surface for the starch. After modification, SP and SP-g-PMAAs exhibited distorted and rougher surfaces which are assumed to be good for adsorption. The dual modification produced phosphate and carboxylic groups on the starch backbone. The phosphate and carboxylic groups have high tendency to make hydrogen bonding with phenol. The comparative adsorption studies of NS, SP and SP-g-PMAAs were checked for phenol. The experimental results showed adsorption efficiencies of 0.022 g/g, 0.082 g/g, 0.100 g/g, 0.148 g/g and 0.151 g/g for NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3, respectively. The following findings revealed that dual modification enhanced the adsorption ability of NS.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Corn starch is well-known as a bulking agent, water retention agent, gelling agent and used as a thickener [1]. On the basis of amylose and amylopectin ratio starch is categorized as high, normal and waxy amylose [2]. Starch is chemically modified by different methods, but dual modification is one of the best ways to get the desire property of the starch [3,4,5,6]. Dually modified starches improve the solubility, rheological stability [7] and increase the tensile strength [8], peak viscosity and swelling property [9]. In the modern world, the cigarette industry is very popular from an economic point of view, but the cigarette is injurious to human health. Cigarette smoke has so many toxic chemical gases [10]. Almost 69 chemicals are well known in cigarette smoke which causes different types of cancer [11,12,13] like lung [14], kidney [15], larynx [16], neck and so on [17]. Phenol is one of the toxic chemicals present in the cigarette smoke [18,19,20]. Phenol is the igniting and burning compound [21] responsible to raise the chances of convulsion and coordinate impairment, muscle weakness and cancer [22,23,24,25]. Various methods are employed for the removal of toxic gases, and adsorption is one of the most effective methods because this is a easy, cheap and environmentally friendly method [26,27,28]. For the removal of phenol, different chemically modified starches were used. Similarly, ginko seed starch spherules were successfully used for the adsorption of phenol and its derivatives, p-chlorophenol and p-nitrophenol [29]. C. Qiu et al. effectively used worm-shaped amylopectin nanoparticles and spherical shaped amylose nanoparticles for the removal of phenol and its derivatives [30]. However, the adsorption efficiency of starch towards phenol needs more attention. In this research article we have prepared novel bio-based SP-g-PMAAs by dual modification. The SP-g-PMAAs was biodegradable, environmentally friendly and cost-effective. The main purpose of dual modification to increase the adsorption efficiency of native starch towards phenol. In first step of synthesis, we coded phosphate groups on the starch main chain. The phosphate groups (acting as hydrogen bond acceptor) were capable to make hydrogen bonding with the hydroxyl group (acting as hydrogen bond donor) of phenol. Thus the introduction of phosphate groups increased the adsorption efficiency of modified starch. The adsorption efficiency was further increased by the grafting of methacrylic acid on the backbone of starch phosphate in the second step of dual modification. The grafting of methacrylic acid increased the density of carboxylic groups (acting as both hydrogen bond donor and acceptor) on starch phosphate. These carboxylic groups made more hydrogen bonding with phenol and grab more phenol. Thus dual modification increased the adsorption efficiency of native starch towards phenol.
Experimental section
Materials
Native starch (NS) was purchased from Aladdin. Phenol (99.0%), ethanol (99.7%), methacrylic acid (MAA) (99.0%), methanol (99.5%) and potassium persulfate (KPS) (99.0%) were bought from Sinopharm Chemical Reagent Co. Ltd., China. Monosodium phosphate (NaH2PO4) (99.0%) and disodium phosphate (Na2HPO4) (99.0%) were bought from J & K Chemical Ltd. All the reagents were used without further purification.
Phosphorylation of starch
The procedure adopted for the phosphorylation of starch was already reported in the literature with some modifications [31, 32]. In this typical procedure, 4.3188 g (36 mmol) of monosodium phosphate and 1.2756 g (8.98 mmol) of disodium phosphate were taken in three necked round bottom flask and added 10 mL of distilled water to dissolve it. During this process the temperature was maintained 35 ˚C. The pH of the reaction system was attuned at 5 by adding drop-wise 3 M NaOH. In next step 5.0059 g (30.96 mmol AGU) of NS was added to the phosphate solution. The reaction mixture was stirred at 450 rpm for 0.5 h to prepare the slurry. To remove the water contents from slurry the mixture was centrifuged. After centrifugation, the half-dried slurry was placed at 40 ˚C for 30 h in vacuum oven. The dried mixture was converted into fine powder by pestle and mortar. The finely powdered mixture was again put in the vacuum oven at 75 ˚C for 2 h. For phosphorylation, the mixture was heated at 150 ˚C for 6.5 h. After 6.5 h, the obtained product was washed three times with water and methanol (1:1). To remove the methanol and water contents the product was washed five times with ethanol. Lastly, the product was placed at 40 ˚C for 24 h in a vacuum oven. The dried starch phosphate (SP) was stored in a desiccator for further characterization and use.
Synthesis of starch phosphate-g-polymethacrylic acids (SP-g-PMAAs)
The methodology adopted for the synthesis of starch phosphate-g-polymethacrylic acids (SP-g-PMAAs) was described in the literature [29]. For all the SP-g-PMAAs, the same reaction conditions were used, except the variant molar ratios of the monomers. For better understanding of the synthesis, SP-g-PMAA3 is taken for an example. Firstly, 1.0453 g (6.45 mmol) of SP was charged into a three-necked flask and 40 mL of distilled water was added to disperse the SP. The reaction was run under argon gas atmosphere at 70 ˚C with constant stirring 450 rpm. After 0.5 h, 278.8 mg (1.03 mmol) of the KPS was dissolved in 10 mL of distilled water and added to the reaction mixture dropwise. Then, 8.2 mL (96.75 mmol) of MAA was introduced to the reaction mixture dropwise and the reaction mixture was stirred for 2 h at the same reaction conditions. After 2 h, the reaction was stopped and cool to room temperature. Afterward, the reaction contents were washed with mixture of methanol (70 mL) and water (30 mL) five times. Finally, the product was washed with ethanol to remove the water and methanol traces. The product obtained after washing was placed at 40 ˚C for 24 h in a vacuum oven. The dried SP-g-PMAA3 was crushed into fine powder by pestle and mortar.
Characterization
The 31P NMR and 1H NMR spectra were recorded on Bruker Advanx-400 DMX NMR spectrometer using D2O (basic). Nicolet 5700 IR spectrometer was used to record FT-IR spectra. The crystalline nature was checked on by using a XPert PRO (Cu Kα, λ = 1.54 Å). Thermal properties were checked on TA-Q500 (Mettler-Toledo) under nitrogen atmosphere. The SEM images were taken on SU-8010 spectrometer (Oxford X-max80 instrument Hitachi Ltd). The Varian CARY 100 Bio UV/Visible spectrophotometer was used for the determination of phenol concentration.
Adsorption experiment
Phenol is regarded as priority pollutant because it is harmful to human even in small amount [33]. There are two types of phenol toxicity: acute and chronic exposure. The acute poisoning of phenol in human can be seen through symptoms such as dryness in throat and mouth, and dark colors of urine excreted due to the occurring of lipid peroxidation [34]. Chronic exposure causes several symptoms such as anderoxia, muscle pain, headache, gastrointestinal pain which may then lead to cancer [35]. Cigarette smoke containing phenol directly contacting human body. Oral exposure to phenol damages to the liver and kidneys. It is reported that 1 g accidental ingestion of phenol may causes fatality to human [36]. So, it is of prior importance to minimize the concentration of phenol in cigarette smoke to make it less harmful to human health. Keeping in view the toxic effects of phenol, SP-g-PMAAs were tested as adsorbents for the removal of phenol. The adsorption experiment was arranged to determine the phenol adsorption affinity of the modified starches.
Filling of sample glass tubes
The samples glass tubes were filled by cutting the cigarette filter into two equal halves. One of the two halves of cigarette filter was put in the sample glass tubes and charged with the specific amount of adsorbent. The other half of the cigarette filter was positioned so that the adsorbent was sandwiched between the two halves. One sample glass tube was filled with blank cigarette filter while the other five tubes were filled with adsorbents. The pictorial procedure for the filling of sample glass tubes is shown in Fig. 1.

Proposed procedure for the filling of sample glass tubes
Phenol adsorption
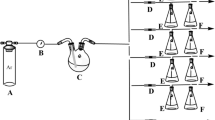
For phenol adsorption experiment the assembly used is shown in Fig. 2. The phenol source solution was prepared in two-necked flask (C) by dissolving 29.0118 g of phenol in a mixture 100 mL of distilled water and 50 mL of ethanol. The two-necked flask was set at 35 ˚C in an oil bath. One neck of the two-necked flask was linked to the gas flow meter (B) which intern connected to source of argon (Ar) gas (A). The second neck of the two-necked flask was attached to the six sample glass tubes (D) connected to six Erlenmeyer flasks (E).

Assembly used for phenol adsorption experiment
The Ar gas was first blown via gas flow meter, and regulated the gas flow at 0.5 m3/h. Then Ar gas was passed via the source solution of phenol where Ar gas obtained phenol as gaseous mixture of Ar/phenol. The gaseous Ar/phenol mixture was blown through the sample glass tubes. One of the six sample glass tubes was labeled blank filled with cigarette filter while the other tubes were charged with 15.1 mg of NS, 15.2 mg of SP, 15.1 mg of SP-g-PMAA1, 15.2 mg of SP-g-PMAA2 and 15.1 mg of SP-g-PMAA3. Here the adsorbents adsorbed certain extent of the phenol and rest of the phenol was let into the ethanol (75 mL) containing Erlenmeyer flasks (E). The ethanol in Erlenmeyer flasks dissolved small amount of phenol. The argon/phenol mixture was blown for 30 min through the sample glass tubes. Then, from each Erlenmeyer flasks (E), 2 mL of ethanol solution was taken and analyzed by UV–visible spectrophotometer to determine the phenol concentration.
Results and discussion
Synthesis and characterization of starch phosphate and SP-g-PMAAs
The SP was synthesized by the reaction of NS with inorganic monosodium phosphate and disodium phosphate salts. Later, SP-g-PMAAs were produced by free radical polymerization of SP with MAA. For the initiation of reaction KPS was used as an initiator. The reaction schemes for the production of SP and SP-g-PMAAs are shown in Scheme 1, while the recipe details for SP-g-PMAAs are listed in Table 1. The SP and SP-g-PMAAs were characterized by different spectroscopic techniques.

Synthesis of SP and SP-g-PMAAs
The 31P NMR spectra of SP, monosodium phosphate and disodium phosphate are shown in Fig. 3. The spectra were taken in basic solution of D2O. The peak appeared at 2.553 ppm was attributed to the unreacted salts. The peak at 1.547 ppm was for the phosphorus atoms labeled by b, e and c. The peaks appeared at 1.108 and -0.012 ppm were assigned to phosphorus atoms labeled as a and f in the glucose units.

31P NMR spectra of (a) SP, (b) monosodium phosphate and (c) disodium phosphate [37]
The structure of SP-g-PMAAs was analyzed by 1H NMR spectrometer. The peak appeared at 5.1–5.3 ppm was assigned to C-1 protons of SP AGU. The peak shifted to downfield because the proton attached to C-1 was highly deshielded. For the protons of C-2 to C-6 of the SP AGU the peaks were broadened from 3.2–4.2 ppm. The two characteristics peaks were appeared at ranged from 1.5–1.7 to 0.8–1 ppm for C-7 and C-8 protons of the PMAA part. The peak for C-7 protons was appeared in upfield because this carbon was directly attached to electronegative oxygen atom. The appearance of peaks for C-7 and C-8 proton confirmed the successful synthesis of SP-g-PMAAs [38]. The 1H NMR spectra for SP-g-PMAAs are shown in Fig. 4. The grafting ratios of PMAA to AGU for SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 were deliberate from 1HNMR.

1H NMR spectra of (a) SP, (b) SP-g-PMAA1 (c) SP-g-PMAA2 and (d) SP-g-PMAA3 [38]
The relative grafting ratios for SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 were calculating by following the Eq. (1).[8]
The calculated grafting values were found to be 0.38 mol/mol, 0.48 mol/mol and 0.53 mol/mol for SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3.
The FT-IR spectra of NS, SP, SP-g-PMAA1, SP-g-PMAA2, and SP-g-PMAA3 are depicted in Fig. 5a. The characteristic peak appeared at 1248 cm−1 was assigned to the stretching vibration of P = O. This peak was absent in NS, which showed the different nature of SP from starch. The peak appeared at 3440 cm−1 was ascribed to the stretching vibration of –OH. Similarly, the peak at 2940 cm−1 was attribute to the stretching vibration of –CH [39]. The characteristics peak appeared at 1716 cm−1 was ascribed to –C = O stretching vibration of the carbonyl group of PMAA [29]. So, that particular peak at 1716 cm−1 showed the successful synthesis of SP-g-PMAA1, SP-g-PMAA2, and SP-g-PMAA3.

(a) FT-IR spectra and (b) XRD patterns of NS, SP, SP-g-PMAA1, SP-g-PMAA2, and SP-g-PMAA3
The XRD patterns of NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 are pictured in Fig. 5b. The starch showed peaks at 2θ = 11.12°, 15.15°, 17.09°, 18.03°, 19.92°, 22.90° and 26.98°. This pattern exhibited the A-type crystalline structure of starch [40]. After phosphorylation all these peaks were disappeared in SP. The loss in crystallinity was due to decreasing of the intra-molecular hydrogen bonding [29]. Similarly, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 exhibited almost amorphous nature.
The TGA and DTG curves for SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 are depicted in Fig. 6. SP showed DTGmax at 239 ˚C and 286 ˚C for the degradation of phosphate group and glycosidic linkages respectively. DTGmax for SP-g-PMAA1 was recorded at 250 ˚C, 296 ˚C and 400 ˚C which were ascribed to thermal decomposition of phosphate group, glycosidic linkage of glucose unit and PMAA constituent respectively. The DTGmax for SP-g-PMAA2 was observed at 246 ˚C, 293 ˚C and 402 ˚C which were attributed to the release of phosphate group, breakdown of glycosidic linkages and PMAA part. Similarly, SP-g-PMAA3 showed DTGmax at 236 ˚C, 281 ˚C and 400 ˚C for the loss of phosphate group, glycosidic linkages and PMAA part, respectively. So, two step weight loss for SP and the three step weight loss for SP-g-PMAAs confirmed the grafting of PMAA onto SP.

(a) TGA and (b) DTG curves of SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3
The surface morphology of the NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 was examined by SEM, respectively. The SEM images showed that starch had spherical or polyhedral shape with smooth surface [41]. After phosphorylation the surface morphology of the NS was changed from the smooth to rough surface. But after the grafting of PMAA onto SP the surface became rougher, which would facilitate the adsorption property [8]. The surface morphology of the NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 are depicted in Fig. 7.

SEM images of (A and B) NS, (C and D) SP, (E and F) SP-g-PMAA1, (G and H) SP-g-PMAA2 and (I and J) SP-g-PMAA3
Adsorption studies
The adsorption experiment was conducted to find out the sorption capability of the SP-g-PMAAs for phenol. In phenol adsorption experiment, the Ar/phenol gaseous mixture was blown via the sample glass tubes. The phenol adsorbed by each adsorbent was calculated by subtracting the phenol concentration in the Erlenmeyer’s flasks (E) connected to the adsorbent containing sample glass tubes from the phenol concentration in the Erlenmeyer’s flasks (E) linked with the blank cigarette filter containing sample glass tubes. The results related to phenol adsorption experiments are shown in Fig. 8.

Adsorption efficiency towards phenol by NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3, respectively after 30 min
The obtained results showed adsorption efficiencies of 0.022 g/g, 0.082 g/g, 0.100 g/g, 0.148 g/g and 0.151 g/g by NS, SP, SP-g-PMAA1, SP-g-PMAA2 and SP-g-PMAA3 for phenol, respectively. The adsorption mechanism for the removal of phenol is given in Scheme 2. The surface of NS saturated with phosphate and carboxylic groups held the phenol by hydrogen bonding. The phosphate and carboxylic groups formed hydrogen bonding with the hydroxyl group of phenol. The possible adsorption process was found to be physi-sorption [8].

Adsorption of phenol by SP-g-PMAAs through hydrogen bonding
Conclusion
In this research, novel dually modified SP-g-PMAAs were synthesized by phosphorylation and free radical polymerization. The main aim of the dual modification was to improve the adsorption ability of the native starch towards phenol. The structures of SP and SP-g-PMAAs were confirmed by 31P NMR, 1H NMR and FT-IR. Due to modification, the crystallinity of the SP-g-PMAAs was diminished for the loss of intera-molecular hydrogen bonding. The phosphorylation generated phosphate groups on the starch main chain. These phosphate groups were capable to grab the phenol by making hydrogen bonding. The adsorption affinity of the starch phosphate towards phenol was further improved by grafting methacrylic acid onto starch phosphate backbone. The grafting of methacrylic acid increased the density of carboxylic groups on starch phosphate which intern adsorb more phenol. The NS, SP, SP-g-PMAA1, SP-g-PMAA2, and SP-g-PMAA3 showed adsorption efficiencies of 0.022 g/g, 0.082 g/g, 0.100 g/g, 0.148 g/g and 0.151 g/g, respectively. The SP-g-PMAA3 showed higher adsorption efficiency towards phenol due to maximum grafting value. The obtained results showed that dual modification of starches by phosphorylation and grafting can increase the adsorption ability of native starch towards phenol. These dually modified starches can be used as additive in cigarette filter to remove phenol from the cigarette smoke.
References
Özmen N (2012) A study of the effect of acetylation on hemp fibres with vinyl acetate. BioResources 7:3800–3809
Singh N, Sandhu KS, Kaur M (2005) Physicochemical properties including granular morphology, amylose content, swelling and solubility, thermal and pasting properties of starches from normal, waxy, high amylose and sugary corn. Progress in Food Biopolymer Research 1:43–55
Raina C, Singh S, Bawa A, Saxena D (2006) Some characteristics of acetylated, cross-linked and dual modified Indian rice starches. Eur Food Res Technol 223(4):561–570. https://doi.org/10.1007/s00217-005-0239-z
Adebowale KO, Afolabi TA, Olu-Owolabi BI (2006) Functional, physicochemical and retrogradation properties of sword bean (Canavalia gladiata) acetylated and oxidized starches. Carbohyd Polym 65:93–101
Haq F, Yu H, Wang L, Teng L, Haroon M, Khan RU, Mehmood S, Ullah RS, Khan A, Nazir A (2019) Advances in chemical modifications of starches and their applications. Carbohyd Res 476:12–35
Huang J, Schols HA, Jin Z, Sulmann E, Voragen AG (2007) Pasting properties and (chemical) fine structure of acetylated yellow pea starch is affected by acetylation reagent type and granule size. Carbohyd Polym 68:397–406. https://doi.org/10.1016/j.carbpol.2006.12.019
Bhandari PN, Singhal RS (2002) Studies on the optimisation of preparation of succinate derivatives from corn and amaranth starches. Carbohyd Polym 47(3):277–283. https://doi.org/10.1016/S0144-8617(01)00202-8
Haroon M, Yu H, Wang L, Ullah RS, Haq F, Teng L (2019) Synthesis and characterization of carboxymethyl starch-g-polyacrylic acids and their properties as adsorbents for ammonia and phenol. Int J Biol Macromol 138:349–358. https://doi.org/10.1016/j.ijbiomac.2019.07.046
Sun F, Liu J, Liu X, Wang Y, Li K, Chang J, Yang G, He G (2017) Effect of the phytate and hydrogen peroxide chemical modifications on the physicochemical and functional properties of wheat starch. Food Res Int 100:180–192. https://doi.org/10.1016/j.foodres.2017.07.001
John E, Coburn S, Liu C, McAughey J, Mariner D, McAdam K, Sebestyén Z, Bakos I, Dóbé S (2018) Effect of temperature and humidity on the gas–particle partitioning of nicotine in mainstream cigarette smoke: A diffusion denuder study. J Aerosol Sci 117:100–117. https://doi.org/10.1016/j.jaerosci.2017.12.015
Branton P, Lu AH, Schüth F (2009) The effect of carbon pore structure on the adsorption of cigarette smoke vapour phase compounds. Carbon 47:1005–1011
Mendel JR, Baig SA, Hall MG, Jeong M, Byron MJ, Morgan JC, Noar SM, Ribisl KM, Brewer NT (2018) Brand switching and toxic chemicals in cigarette smoke: A national study. PLoS ONE 13:e0189928
Noar SM, Kelley DE, Boynton MH, Morgan JC, Hall MG, Mendel JR, Ribisl KM, Brewer NT (2018) Identifying principles for effective messages about chemicals in cigarette smoke. Prev Med 106:31–37
Tindle HA, Duncan MS, Greevy RA, Vasan RS, Kundu S, Massion PP, Freiberg MS (2018) Lifetime smoking history and risk of lung cancer: Results from the framingham heart study. J Natl Cancer Inst 110:1201–1207
Chow WH, Dong LM, Devesa SS (2010) Epidemiology and risk factors for kidney cancer. Nature Reviews Urology 7:245–257
Zuo JJ, Tao ZZ, Chen C, Hu ZW, Xu YX, Zheng AY, Guo Y (2017) Characteristics of cigarette smoking without alcohol consumption and laryngeal cancer: Overall and time-risk relation. A meta-analysis of observational studies, European Archives of Otorhinolaryngology 274:1617–1631
Beynon RA, Lang S, Schimansky S, Penfold CM, Waylen A, Thomas SJ, Pawlita M, Waterboer T, Martin RM, May M (2018) Tobacco smoking and alcohol drinking at diagnosis of head and neck cancer and all-cause mortality: Results from head and neck 5000, a prospective observational cohort of people with head and neck cancer. Int J Cancer 143:1114–1127
Guo L, Zhu Y, Du X (2012) The effect of modified starches on the adsorption of cigarette mainstream smoke composition. Starch-Stärke 64:552–562
Li G, Yu H, Xu L, Ma Q, Chen C, Hao Q, Qian Y (2011) General synthesis of carbon nanocages and their adsorption of toxic compounds from cigarette smoke. Nanoscale 3:3251–3257
Wang Y, Li Z, Ding J, Hu Z, Liu Z, Zhou G, Huang T (2017) Research on the ordered mesoporous silica for tobacco harm reduction. IOP Conference Series: IOP Publishing 281(1):012018
Berardinelli S, Resini C, Arrighi L (2008) Technologies for the removal of phenol from fluid streams: A short review of recent developments. J Hazard Mater 160:265–288. https://doi.org/10.1016/j.jhazmat.2008.03.045
Ahmaruzzaman M (2008) Adsorption of phenolic compounds on low-cost adsorbents: A review. Adv Coll Interface Sci 143:48–67. https://doi.org/10.1016/j.cis.2008.07.002
Chortyk OT, Schlotzhauer WS (1989) The contribution of low tar cigarettes to environmental tobacco smoke. J Anal Toxicol 13(3):129–134. https://doi.org/10.1093/jat/13.3.129
Michałowicz J, Duda W (2007) Phenols-sources and toxicity. Polish Journal of Environmental Studies 16:347–362
Pistonesi MF, Di Nezio MS, Centurión ME, Lista AG, Fragoso WD, Pontes MJ, Araújo MC, Band BSF (2010) Simultaneous determination of hydroquinone, resorcinol, phenol, m-cresol and p-cresol in untreated air samples using spectrofluorimetry and a custom multiple linear regression-successive projection algorithm. Talanta 83(2):320–323. https://doi.org/10.1016/j.talanta.2010.09.027
Asfaram A, Ghaedi M, Hajati S, Goudarzi A, Dil EA (2017) Screening and optimization of highly effective ultrasound-assisted simultaneous adsorption of cationic dyes onto Mn-doped Fe3O4-nanoparticle-loaded activated carbon. Ultrason Sonochem 34:1–12. https://doi.org/10.1016/j.ultsonch.2016.05.011
Bée A, Obeid L, Mbolantenaina R, Welschbillig M, Talbot D (2017) Magnetic chitosan/clay beads: A magsorbent for the removal of cationic dye from water. J Magn Magn Mater 421:59–64. https://doi.org/10.1016/j.jmmm.2016.07.022
Vanamudan A, Pamidimukkala P (2015) Chitosan, nanoclay and chitosan-nanoclay composite as adsorbents for Rhodamine 6G and the resulting optical properties. Int J Biol Macromol 74:127–135. https://doi.org/10.1016/j.ijbiomac.2014.11.009
Chen Q, Yu H, Wang L, Abdin ZU, Yang X, Wang J, Zhou W, Zhang H, Chen X (2016) Synthesis and characterization of amylose grafted poly (acrylic acid) and its application in ammonia adsorption. Carbohyd Polym 153:429–434. https://doi.org/10.1016/j.carbpol.2016.07.120
Qiu C, Qin Y, Zhang S, Xiong L, Sun Q (2016) A comparative study of size-controlled worm-like amylopectin nanoparticles and spherical amylose nanoparticles: Their characteristics and the adsorption properties of polyphenols. Food Chem 213:579–587. https://doi.org/10.1016/j.foodchem.2016.07.023
Passauer L, Liebner F, Fischer K (2009) Starch phosphate hydrogels. Part I: Synthesis by mono-phosphorylation and cross-linking of starch, Starch - Stärke 61:621–627
Pourjavadi A, Doulabi M, Soleyman R, Sharif S, Eghtesadi SA (2012) Synthesis and characterization of a novel (salep phosphate)-based hydrogel as a carrier matrix for fertilizer release. React Funct Polym 72(10):667–672. https://doi.org/10.1016/j.reactfunctpolym.2012.06.010
Agostini E, Talano MA, González PS, Oller ALW, Medina MI (2011) Phytoremediation of phenolic compounds: Recent advances and perspectives. In Handbook of Phytoremediation, Nova Science Publishers, New York, pp 1–50
Olujimi O, Fatoki O, Odendaal J, Okonkwo J (2010) Endocrine disrupting chemicals (phenol and phthalates) in the South African environment: A need for more monitoring. Water Sa 36:671–682
Chakraborty S, Bhattacharya T, Patel T, Tiwari K (2010) Biodegradation of phenol by native microorganisms isolated from coke processing wastewater. J Environ Biol 31:293–296
Nuhoglu A, Yalcin B (2005) Modelling of phenol removal in a batch reactor. Process Biochem 40:1233–1239. https://doi.org/10.1016/j.procbio.2004.04.003
Shukri R, Shi YC (2017) Structure and pasting properties of alkaline-treated phosphorylated cross-linked waxy maize starches. Food Chem 214:90–95. https://doi.org/10.1016/j.foodchem.2016.07.036
Shalviri A, Chan HK, Raval G, Abdekhodaie MJ, Liu Q, Heerklotz H, Wu XY (2013) Design of pH-responsive nanoparticles of terpolymer of poly(methacrylic acid), polysorbate 80 and starch for delivery of doxorubicin. Colloids Surf, B 101:405–413. https://doi.org/10.1016/j.colsurfb.2012.07.015
Hebeish A, Higazy A, El-Shafei A, Sharaf S (2010) Synthesis of carboxymethyl cellulose (CMC) and starch-based hybrids and their applications in flocculation and sizing. Carbohyd Polym 79(1):60–69. https://doi.org/10.1016/j.carbpol.2009.07.022
da Lazzarotto SRS, Bet C, Hornung P, Lazzarotto M, Schnitzler E (2017) Induced effects by oxidation with potassium permanganate on the thermal, morphological, colorimetric and pasting properties of corn starch. Ukrainian Food Journal 6(2):197–210. https://doi.org/10.24263/2304-974X-2017-6-2-3
Li MC, Lee J, Cho U (2012) Synthesis, characterization, and enzymatic degradation of starch-grafted poly(methyl methacrylate) copolymer films. J Appl Polym Sci 125(1):405–414. https://doi.org/10.1002/app.35620
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Haq, F., Yu, H., Wang, L. et al. Dual modification of starches by phosphorylation and grafting and their application as adsorbents for the removal of phenol. J Polym Res 27, 313 (2020). https://doi.org/10.1007/s10965-020-02280-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-020-02280-5
Keywords
Article history
Latest
Dual modification of starches by phosphorylation and grafting and their application as adsorbents for the removal of phenol- Published:
- 28 September 2020
- Received:
- 17 May 2020
- Accepted:
- 03 September 2020
DOI: https://doi.org/10.1007/s10965-020-02280-5