
Abstract
The structural, elastic, electronic and magnetic properties of the Mn2RhSi Mn2-based Heusler alloy were investigated by using first-principles calculations of the full-potential linearized augmented plane wave (FP-LAPW) method. The results of electronic structures show that the Mn2RhSi exhibits a half-metallic character for a wide range 5.65 to 5.91 Å of lattice constant. The half-metallic behavior is confirmed by the integral value of 3 μ B of the total magnetic moment for Mn2RhSi, which follow the Slater-Pauling rule.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The Heusler alloys have attracted growing research interest because of their potential application as functional materials [1–3]. Numerous half-metallic Heusler alloys have been proposed using first-principles calculations, which are now known to exhibit a shape memory with interesting properties, where one of the most interesting of these materials is the Mn2-based Heusler alloys. The half-metallic ferromagnetism has been discovered in several materials, like the ferromagnetic metallic oxides, dilute magnetic semiconductors, binary transition metal pnictides, chalcogenides with zinc blende structure, and Heusler compounds [4].
The Heusler alloys are important materials with respect to their interesting properties such as the electronic localization, itinerant magnetism, antiferromagnetism, helimagnetism, Pauli paramagnetism, or heavy-fermion behavior [5–8], and among these, the half-metallic property in Mn-based Heusler alloys is discovered in some theoretical and experimental studies. The half-metallic behavior was studied both theoretically [9–11] and experimentally [12] in Mn2VAl. Besides, the half-metallicity is investigated in some Mn2-based Heusler alloys such as Mn2VZ (Z = Si, Al) [13], Mn2CoSn [14], Mn2CoZ (Z = Al, Ga, In, Si, Ge, Sn, Sb) [15], Mn2YAl (Y = V, Fe, Co) [16], Mn2CoSb [17], and Mn2FeZ (Z = Al, Ga, Si, Ge, Sb) [18] and in ferromagnetic shape memory alloys such as Mn2NiGa [19] and Mn2NiZ (Z = In, Sn, Sb) [20]. Although several studies have been devoted for the half-metallic ferrimagnets [21–29], the study of Wei et al. [25], for example, showed that the Mn(1) and Mn(2) atoms in the Mn2CuGe alloy have large antiparallel spin moments with ferrimagnetic behavior. These materials are much more desirable than their ferromagnetic counterparts in magneto-electronic applications [30, 31]. Recently, the half-metallicity has been theoretically predicted for many of Mn2-based Heusler alloys [31–33].
To the best of our knowledge, there are no most works appeared on the electronic and magnetic properties of Mn2RhSi Heusler alloy and there were no studies on the elastic properties of this compound. The purpose of this study is to predict the structural parameters, elastic constants, mechanical stability, electronic structure, and magnetic properties of Mn2RhSi Heusler alloy. The calculations are carried out by using the first-principles calculations of the full-potential linearized augmented plane wave (FP-LAPW) method.
2 Method of Calculations
The electronic and magnetic properties are performed with the FP-LAPW method implemented in Wien2k code [34]. The generalized gradient approximation (GGA) [35] is used for the exchange and correlation potential. The parameter R MT × K max (the smallest muffin-tin radius multiplied by the maximum k value in the expansion of plane waves in the basis set), which controls the size of the basis set in our calculations, is chosen to be equal to 8. The muffin-tin (MT) radii are chosen to be equal to 2.2, 2.3 and 2.06 a.u. for Mn, Rh, and Si, respectively. The basis set is expanded, within these spheres, with the charge density and potential in terms of the spherical harmonics up to the angular momentum l max = 10. The Brillouin zone integrations for the total energy are carried out using 104 special kpoints in the irreducible Brillouin zone. The self-consistent convergence of the total energy was set at 0.1 mRy.
3 Results and Discussions
3.1 Structural Parameters, Elastic Properties, and Mechanical Stability
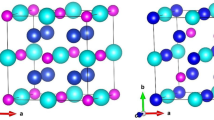
The X2YZ Heusler alloys are ternary compounds, where X and Y are transition metals and Z is a group II, IV or V element [4]. The X2YZ Heusler alloys crystallize into two possible structures: the Cu2MnAl-type structure and the Hg2CuTi-type structure. In the cubic Cu2MnAl-type structure with the space group \(Fm\overline {3}m\) (No. 225) the atoms are located at the Wyckoff coordinates: the X atoms occupy A (0, 0, 0) and B (1/2, 1/2, 1/2) sites, the Y atoms situated at the C site (1/4, 1/4, 1/4) and the Z atom situated at D site (3/4, 3/4, 3/4). In the Hg2CuTi-type structure with the space group \(F\overline {4}3m\) (No. 216), the X atoms occupy A (0, 0, 0) and B (1/4, 1/4, 1/4) positions while the Y and Z atoms are found in C (1/2, 1/2, 1/2) and D (3/4, 3/4, 3/4) positions, respectively [14] (see Fig. 1).

The Mn2RhSi Heusler alloy in the Hg2CuTi-type structure with the atomic positions of Mn1 (0, 0, 0), Mn2 (1/4, 1/4, 1/4), Rh (1/2, 1/2, 1/2), and Si (3/4, 3/4, 3/4)
The stability of the Mn2RhSi compound in the Cu2 MnAl- or Hg2CuTi-type structures must be known beforehand in order to calculate its structural properties. For Mn2RhSi in the two Cu2MnAl- and Hg2CuTi-type structures, the lattice parameters are fully optimized at zero pressure as shown in Fig. 2. From Table 1, it can be seen that the equilibrium energy of the Cu2MnAl-type structure is higher than that of the Hg2CuTi-type, which indicates that Mn2RhSi is energetically more stable in the Hg2CuTi-type structure. This verifies the site preference rule of X and Y atoms, which is strongly affected by the number of their 3d electrons [16].

Variation of total energy as a function of the volume (V) unit cell of Mn2RhSi in the Cu2MnAl- and Hg2CuTi-type structures
The calculated structural parameters of Mn2RhSi in the Hg2CuTi-type structure such as the lattice constant (a), bulk modulus (B), and its pressure derivative (B ′) with the available theoretical data [33] are listed in Table 2. The equilibrium lattice constant of Mn2RhSi is 5.761, which is in agreement with the theoretical parameter found by Ren et al. [33].
The mechanical properties, such as the elastic constants (C 11, C 12, and C 44) and the Zener anisotropy factor (A) for Mn2RhSi, were also calculated. The computed results are summarized in Table 3. Unfortunately, to our knowledge, no other results are available in the literature for comparison. In the present work, the elastic constants C 11, C 12, and C 44 for the Mn2RhSi were calculated by using the applied strains to be volume-conserving. To verify the structural stability of our compound, we check the conditions for the mechanical stability criterion for cubic crystal [36–38]
Our results verify these conditions, indicating clearly that the Mn2RhSi is stable in the Hg2CuTi-type structure.
The Zener anisotropy factor (A) is an important parameter that measures the anisotropy of elastic wave velocity in a crystal structure; it is defined in the following expression [39]:
The anisotropy factors (A) for Mn2RhSi verify the condition (A > 1), which implies that this compound is more rigid in the diagonal direction <111> [40].
3.2 Electronic Properties
We have used the equilibrium lattice constant in the Hg2CuTi-type to calculate the electronic and magnetic properties of Mn2RhSi Heusler alloy. The spin-polarized band structures along high symmetry directions in the Brillouin zone for Mn2RhSi are shown in Fig. 3. The minority-spin bands have an indirect bandgap along the Γ–X direction, showing a semiconducting character at the Fermi level, while the majority-spin bands depicted an intersection between valence and conduction bands at the Fermi level, indicating a metallic nature of the majority-spin channel. Thus, the Mn2RhSi shows a half-metallic behavior.

Spin-polarized band structures for Mn2RhSi at equilibrium lattice constant: a majority spin (up) and b minority spin (down)
To further elucidate the nature of the band structure of our material, their local density of state (LDOS) and total DOS (TDOS) were calculated. Figures 4 and 5 display respectively the trend of the total and partial densities of states for Mn2RhSi. The TDOS depicts that majority-spin states are metallic, whereas the minority-spin states show the existence of a gap of 0.36 eV. Thus, the Mn2RhSi behaves in metallic nature for majority spin and a semiconductor for minority spin, inducing a perfect 100 % spin polarization at the Fermi level (E F). This is due to the high value of the TDOS of spin-up at E F. From Fig. 4 of TDOS, one can notice the existence of three peaks between −3 and 1 eV for the majority spin, which described the bonding and antibonding states of Mn atoms at E F. For the minority spin, the gap stems from the large exchange splitting of the two Mn1 and Mn2 atoms.

Spin-polarized total density of states for Mn2RhSi at equilibrium lattice constant

Spin-polarized partial densities of states for Mn2RhSi at equilibrium lattice constant
Furthermore, the majority-spin valence band is mainly dominated by the d-states of Mn1 and Mn2 atoms that hybridize with d (Rh) and p (Si) states as shown in Fig. 5. On the other hand, the conduction band is principally formed by the hybridization between d (Mn1) and d (Mn2) states. The d (Mn) states of majority spin shifted to higher energies and cross the Fermi level, while in the minority spin, a bandgap appears at E F, between the occupied d (Mn) bonding states and the unoccupied d (Mn) antibonding states. This leads to a bandgap at E F in the minority-spin states and a metallic character at E F in the majority-spin states, which means that Mn2RhSi is half-metal with spin polarization of 100 %. Therefore, the Mn2RhSi material seems to be a potential candidate for spintronic applications.
It is well known that the lattice constant can undergo a considerable variation due to strain and thus deviates from its equilibrium value. This is widely influenced by the half-metallic property for spintronic applications. It is therefore useful to study the variation of the half-metallicity of Mn2RhSi with respect to the lattice constant. However, the half-metallic behavior of our compound is now examined when the lattice constant is changed. For this purpose, the spin-polarized total density of states of Mn2RhSi Heusler alloy is calculated for the different values of the lattice parameter in the range of 5.65 to 6.01 Å. Figure 6 illustrates the TDOS of Mn2RhSi as a function of the lattice constant. The whole structure of the TDOS shifted to higher energies towards the Fermi level. The majority-spin states exhibit only minor changes in shape as the lattice constant varies, but the position of the DOS of minority spin shifted towards a higher energy with increasing of the lattice constant (a) and cross the Fermi level when a = 6.01 Å. Thus, the Mn2RhSi Heusler alloy is half-metal in the range 5.65 to 5.91 Å of the lattice constant, but this compound reveals a metallic behavior at the lattice constant equal to 6.01 Å.

Spin-polarized total densities of states for Mn2RhSi in the range 5.65 to 6.01 Å of lattice constant
3.3 Magnetic Properties
The calculated total and partial magnetic moments of Mn2RhSi Heusler alloy are summarized in Table 4. The computed total magnetic moment is compared to the total magnetic moment with respect to the Slater-Pauling rule. The total magnetic moment (M t) at the equilibrium lattice parameter for Mn2RhSi is an integral number of 3 μ B, which follows the Slater-Pauling rule of M t = Z t − 24 = 3 μ B, where Z t is the number of valence electrons [13].
Our calculations show that Rh atoms have positive magnetic moments, whereas the Mn atoms have antiparallel spin moments. As is often the case, when these transition metals are very close to each other, there is an antiparallel coupling in space. Thus, there is a strong correlation between the lattice parameter and the interaction between magnetic spins of atoms. However, the magnetic alignment of Mn1 and Mn2 atoms is antiparallel with magnetic moments of −0.409 μ B of Mn1 and 3.013 μ B of Mn2. As a consequence, the Mn2RhSi exhibits a half-metallic ferrimagnetic behavior.
4 Conclusion
In summary, we have investigated the structural, elastic, electronic and magnetic properties of the Mn2RhSi of Heusler alloy, using the firstprinciples calculations of the fullpotential linearized augmented plane wave (FP-LAPW) method. From structural and elastic properties, we found that this compound is stable in the Hg2CuTi-type structure. The Mn2RhSi exhibits a half-metallic ferrimagnetic character at equilibrium lattice constant, and this behavior results from the antiparallel magnetic alignment of Mn1 and Mn2 atoms. The total magnetic moment of Mn2RhSi is an integral Bohr magneton of 3 μ B, which obeys to the Slater-Pauling rule and confirms the half-metallicity of this material. From our findings, we have predicted that Mn2RhSi Heusler alloy is a potential candidate for practical spintronic applications.
References
Julliere, M.: Phys. Lett. A 54, 225 (1975)
Jimbo, M., Kanda, T., Goto, S.: J. Magn. Magn. Mater. 126, 422 (1993)
Ohno, H.: Science 281, 951 (1998)
Heusler, F., Deut, V.: Phys. Ges. 5, 219 (1903)
Webster, P.J., Ziebeck, K.R.A.: Alloys and compounds of d-elements with main group elements. Part 2. In: Wijn, H.R.J. (ed.) Landolt-Börnstein, New Series, Group III, Pt.c, vol. 19, p. 75184. Springer, Berlin
Pierre, J., Skolozdra, R.V., Tobola, J., Kaprzyk, S., Hordequin, C., Kouacou, M.A., Karla, I., Currat, R., Leliévre-Berna, E.: J. Alloys Compd. 262, 101 (1997)
Tobola, J., Pierre, J.: J. Alloys Compd. 296, 243 (2000)
Hichour, M., Rached, D., Khenata, R., Rabah, M., Merabet, M., Reshak, A.H., Bin Omran, S., Ahmed, R.: J. Phys. Chem. Solids 73, 975 (2012)
Liu, G.D., Dai, X.F., Yu, S.Y., Zhu, Z.Y., Chen, J.L., Wu, G.H., Zhu, H., Xiao, J.Q.: Phys. Rev. B 74, 054435 (2006)
Weht, R., Pickett, W.E.: Phys. Rev. B 60, 13006 (1999)
Ishida, S., Asano, S., Ishida, J.: J. Phys. Soc. Jpn. 53, 2718 (1984)
Itoh, H., Nakamichi, T., Yamaguchi, Y., Kazama, N.: Trans. Jpn. Inst. Met. 24, 265 (1983)
Wurmehl, S., Kandpal, H.C., Fecher, G.H., Felser, C.: J. Phys. Condens. Matter 18, 6171 (2006)
Al-zyadi, J.M.K., Gao, G.Y., Yao, K.L.: J. Alloys Compd. 565, 17 (2013)
Liu, G.D., Dai, X.F., Liu, H.Y., Chen, J.L., Li, Y.X., Xiao, G., Wu, G.H.: Phys. Rev. B 77, 014424 (2008)
Luo, H., Zhu, Z., Ma, L., Xu, S., Zhu, X., Jiang, C., Xu, H., Wu, G.: J. Phys. D Appl. Phys. 41, 055010 (2008)
Dai, X.F., Liu, G.D., Chen, J.L., Wu, G.H.: Solid State Commun. 140, 533 (2006)
Luo, H.Z., Zhang, H.W., Zhu, Z.Y., Ma, L., Xu, S.F., Wu, G.H., Zhu, X.X., Jiang, C.B., Xu, H.B.: J. Appl. Phys. 103, 083908 (2008)
Barman, S.R., Chakrabarti, A.: Phys. Rev. B 77, 176401 (2008)
Luo, H., Liu, G., Feng, Z., Li, Y., Ma, L., Wu, G., Zhu, X., Jiang, C., Xu, H.: J. Magn. Magn. Mater. 321, 4063 (2009)
Fujii, S., Okada, M., Ishida, S., Asano, S.: J. Phys. Soc. Jpn. 77, 74702 (2008)
Luo, H.Z., Zhu, Z.Y., Liu, G.D., Xu, S.F., Wu, G.H., Liu, H.Y., Qu, J.P., Li, Y.X.: J. Magn. Magn. Mater. 320, 421 (2008)
Xing, N.S., Li, H., Dong, J.M., Long, R., Zhang, C.W.: Comp. Mater. Sci. 42, 600 (2008)
Luo, H., Liu, G., Feng, Z., Li, Y., Ma, L., Wu, G., Zhu, X., Jiang, C., Xu, H.: J. Magn. Magn. Mater. 321, 4063 (2009)
Wei, X.P., Hu, X.R., Mao, G.Y., Chu, S.B., Lei, T., Hu, L.B., Deng, J.B.: J. Magn. Magn. Mater. 322, 3204 (2010)
Luo, H.Z., Meng, F.B., Feng, Z.Q., Li, Y.X., Zhu, W., Wu, G.H., Zhu, X.X., Jiang, C.B., Xu, H.B.: J. Appl. Phys. 105, 103903 (2009)
Wei, X.P., Deng, J.B., Chu, S.B., Mao, G.Y., Hu, L.B., Yang, M.K., Hu, X.R.: Comp. Mater. Sci. 50, 1175 (2011)
Luo, H.Z., Liu, G.D., Meng, F.B., Wang, L.L., Liu, E.K., Wu, G.H., Zhu, X.X., Jiang, C.B.: Comp. Mater. Sci. 50, 3119 (2011)
Wei, X.P., Hu, X.R., Liu, B., Lei, Y., Deng, H., Yang, M.K., Deng, J.B.: J. Magn. Magn. Mater. 323, 1606 (2011)
Pickett, W.E., Moodera, J.S.: Phys. Today 54, 39 (2001)
Zenasnia, H., Faraoun, H.I., Esling, C.: J. Magn. Magn. Mater. 333, 162 (2013)
Bensaid, D., Hellal, T., Ameri, M., Azzaz, Y., Doumi, B., Al-Douri, Y., Abderrahim, B., Benzoudji, F.: J. Supercond. Nov. Magn. 29, 1843 (2016)
Ren, Z., Liu, Y., Li, S., Zhang, X., Liu, H.: Mater Sci-Poland. doi:10.1515/msp-2016-0043
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2k, An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. Vienna University of Technology, Vienna (2001)
Perdew, J.P., Burke, K., Ernzerhof, M.: Phys. Rev. Lett. 77, 3865 (1996)
Mehl, M.J., Osburn, J.E., Papaconstantopoulos, D.A., Klein, B.M.: Phys. Rev. B 41, 10311 (1990)
Mehl, M.J.: Phys. Rev. B 47, 2493 (1993)
Wang, J., Yip, S., Phillpot, S.R., Wolf, D.: Phys. Rev. Lett. 71, 4182 (1993)
Nye, F.J.: Physical Properties of Crystals. Oxford University Press, Oxford (1985)
Karki, B.B., Ackland, G.J., Crain, J.: J. Phys. Condens. Matter 9, 8579 (1997)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zemouli, M., Boudali, A., Doumi, B. et al. First-Principles Investigation of Elastic, Electronic, and Half-Metallic Ferrimagnetic Properties in the Mn 2 RhSi Heusler Alloy. J Supercond Nov Magn 29, 3187–3192 (2016). https://doi.org/10.1007/s10948-016-3719-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-016-3719-4