
Abstract
Tau is a microtubule-associated protein that is mainly expressed in central and peripheral nerve systems. Tau binds to tubulin and regulates assembly and stabilization of microtubule, thus playing a critical role in neuron morphology, axon development and navigation. Tau is highly stable under normal conditions; however, there are several factors that can induce or promote aggregation of tau, forming neurofibrillary tangles. Neurofibrillary tangles are toxic to neurons, which may be related to a series of neurodegenerative diseases including Alzheimer’s disease. Thus, tau is widely accepted as an important therapeutic target for neurodegenerative diseases. While the monomeric structure of tau is highly disordered, the aggregate structure of tau is formed by closed packing of β-stands. Studies on the structure of tau and the structural transition mechanism provide valuable information on the occurrence, development, and therapy of tauopathies. In this review, we summarize recent progress on the structural investigation of tau and based on which we discuss aggregation inhibitor design.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tau is a microtubule-associated protein that is mainly expressed in central and peripheral nerve systems. Tau binds to tubulin and regulates assembly and stabilization of microtubule, thus playing a critical role in neuron morphology, axon development and navigation [1, 2]. In addition to regulating microtubule assembly, recent studies show that tau has other functions [3,4,5,6]. For example, tau regulates the function of mitochondria, dynamics of RNA, formation of stress granules, integrity of neuronal DNA, motility of motor proteins, and the signaling pathway of brain insulin [7,8,9,10,11,12].
Under normal conditions, wild type tau protein is highly soluble, showing little tendency for aggregation; however, under pathological conditions, a variety of factors have been shown to induce or promote tau aggregation, including mutation, post-translational modification (PTM), metal ions, and interaction with polyanion or other molecules. Aggregation of tau into neurofibrillary tangles (NFTs) characterizes a series of neurodegenerative diseases termed as tauopathies, including Alzheimer’s disease (AD), Parkinson’s disease (PiD), Huntington’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), agyrophilic grain disease, and frontotemporal dementia with parkinsonism‑17 [13, 14]. Consequently, tau is widely considered as a potential target for the treatment of tauopathies [15,16,17,18,19,20].
So far, several strategies have been applied to reduce tau aggregation [2, 15,16,17,18,19,20,21]. Aggregation inhibitors directly bind to tau and block its aggregation. Molecules stabilizing microtubules enhance binding of tau to microtubules, thus reducing the concentration of free tau. Molecules targeting pathways involved in tau hyperphosphorylation or acetylation also suppress tau aggregation by reducing tau PTMs. Furthermore, anti-tau vaccines, via active or passive immunotherapies, enhance clearance of tau aggregates. In this review, we summarize recent progress on the structural characterization of tau, based on which tau targeted aggregation inhibitor design/discover is discussed.
2 Primary Structure of Tau
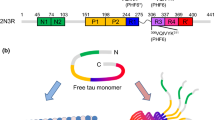
Human tau protein (UniProt ID P10636) is encoded by the MAPT gene which is located on chromosome 17q21 and comprises 16 exons [22]. Alternative splicing of exons 2, 3 and 10 generates six different tau isoforms, which contain zero, one or two N-terminal inserts (0 N, 1 N and 2 N) and three or four microtubule-binding repeats (3R and 4R) (Fig. 1a) [23, 24]. The amounts of 3R and 4R tau isoforms are approximately equal in normal brain [25], but their distributions can be uneven in tauopathies. Based on the biochemical properties and functions, the tau protein can be divided into four distinct domains: the N-terminal domain (NTD), the proline-rich domain (PRD), the microtubule-binding domain (MTBD), and the C-terminal domain (CTD) (Fig. 1a) [1, 26, 27]. The NTD and PRD form the projection domain which extends outward from the microtubule surface when tau associates with microtubule via the MTBD [28, 29]. Compared to 3R tau, 4R tau contains one more repeat. Consequently, 4R tau exhibits higher affinity for microtubule and promotes microtubule assembly more efficiently [2, 25]. As shown in Fig. 1b, the amino acid sequence of 2N4R tau is mostly hydrophilic, low complexity, and locally repetitive. 4R tau contains two aggregation-prone hexapeptide motifs, PHF6* (275VQIINK280) and PHF6 (306VQIVYK311), as well as two cysteine residues, Cys291 and Cys322. PHF6* and Cys291 are located on the R2 repeat of MTBD while PHF6 and Cys322 are located on the R3 repeat. PHF6 and PHF6* are critical for tau aggregation. Tau molecules lacking these two hexapeptide motifs cannot aggregate [30]. The two cysteine residues also regulate tau aggregation in a DTT dependent manner. In the presence of DTT, cysteine to alanine mutation delays the initial aggregation kinetics [31]. On the contrary, cysteine to alanine mutation promotes aggregation in the absence of DTT [32]. Charge distribution analysis reveals that the NTD and CTD are mainly negatively charged (Fig. 1c). On the contrary, the PRD and MTBD are highly positively charged. As discussed below, such a charge segregation feature has a critical impact on the structure and function of tau.

Primary structure of tau protein. a Human MAPT gene encodes six tau isoforms that are resulted from alternative splicing of exons 2, 3, and 10. N1 and N2 are the N-terminal inserts. P1 and P2 are the two proline rich regions. R1 to R4 are the four microtubule-binding repeats. R’ is the C-terminal repeat-like region. b Amino acid sequence of 2N4R tau isoform. Individual region is indicated by the same color as in (a). The two hexapeptide motifs and the two cysteine residues are highlighted in yellow. c Net charge per residue (NCPR) distribution of 2N4R tau. NCPR was analyzed by CIDER [182]. d Residue type specific post-translational modifications of tau (Color figure online)
Tau can be subjected to a large number of PTMs (Fig. 1d) [33,34,35]. 2N4R tau contains 45 serine residues, 35 threonine residues, and 5 tyrosine residues. Within these 85 potential phosphorylation sites, phosphorylation on about 45 sites has been observed experimentally [36]. It has been found that phosphorylation plays a critical role in regulating the function of tau, and the phosphorylation state of tau is developmentally regulated [36]. Fetal tau and adult tau carry approximately seven and two phosphates per molecule on average, respectively [37]. Although the phosphorylation extent of tau is low in normal adults, it rises again in AD patients, with eight phosphates per molecule on average [38], indicating a correlation between tau phosphorylation and tauopathies [39, 40]. Besides phosphorylation, acetylation is another major PTM regulating the function and stability of tau. Tau contains 44 lysine residues and more than twenty of them can be acetylated [41,42,43]. Tau acetylation is detected in various transgenic mice and human tauopathies, suggesting that tau acetylation could be involved in the pathogenesis of neurodegenerative diseases [44, 45]. Similar to phosphorylation, acetylation can also impair tau–microtubule interactions and result in tau aggregation [41, 44,45,46,47,48,49]. Acetylation and phosphorylation may compete with each other or one PTM may promote the other [49, 50]. Ubiquitination usually drives protein degradation. It is surprised to find that tau filaments from CBD and AD brain tissues are ubiquitinated and ubiquitination of tau mediates the filament structures [51]. Recently, Kametani et al. investigated PTMs of tau associated with a wide range of tauopathies [52]. They identified 170 PTMs in total, among which disease-specific PTMs are usually found in the MTBD. Theses disease-specific PTMs may contribute to form the filaments, or they may occur after the filaments have been formed.
3 Secondary Structure of Tau
Although tau forms extensive β-strands in the aggregated filaments, free tau monomer is intrinsically disordered, with low propensity of forming secondary structures (Fig. 2a) [53]. Circular dichroism (CD) confirms that tau has very little secondary structure [54, 55]. The α-helix and β-strand contents are estimated to be less than 5% and 15%, respectively [55]. Nevertheless, nuclear magnetic resonance (NMR) reveals that dynamic and residual secondary structures are present in tau monomer [56,57,58,59,60] (Fig. 2b). Segments showing β-structure conformations include 86GKQAAAQ92 (in N2), 161GQKGQA166 (in P1), 224KKVAVVR230 (in P2), 256VKSKIG262 (in R1), 274KVQIINKKLDL284 (in R2), 305SVQIVYKPVDL315 (in R3), 336QVEVKSEKLD345 and 351QSKIGSL357 (in R4), where segments comprising PHF6* and PHF6 exhibit the highest β-structure propensity [57]. α-Helical propensity is observed for 114LEDEAAGHVT123 (between N2 and P2) and 428LADEVSASLA437 (in CTD). Polyproline II helical conformation is identified for three segments within the PRD, i.e., 175TPPAPKTPPS184 (in P1), 216PTPPTREP223 and 232PPKSPSSA239 (in P2). The formation of these secondary structure elements is transient. For example, the β-structure conformation is populated 12% of the time for 256VKSKIG262 segment and the α-helical conformation is populated 25% of the time for 428LADEVSASLA437 segment [57].

Secondary structure of tau protein. a Secondary structure propensity of 2N4R tau predicted by PSSpred [183]. b Transient secondary structure elements in 2N4R tau identified from NMR characterization: β-structure (red), α-helical (green), polyproline II (blue) (Color figure online)
Solvent conditions, mutations, PTMs, metal ions as well as intermolecular interactions can remodel the conformational propensity of tau monomer. In the presence of 50% trifluoroethanol, the α-helix content of tau rises to 30% [55]. Inferred from NMR data, the 301P-K311 segment forms nascent β-structure, where a type II β-turn is formed around 301PGGG304 [59]. It has been found that Pro301 is critical in maintaining this β-hairpin since P301L mutation promotes conversion from collapsed hairpin to extended conformations [61]. C291R, a mutation with potential pathogenic function, is found to enhance the β-structure propensity of MTBD [62]. 2N4R tau contains two cysteine residues and twelve histidine residues, which provide coordination sites for metal ions. For example, tau binds Zn2+ and becomes more compacted globally [63]; however, structural changes around the MTBD may be subtle [64]. Tau interacts with various polyanions, such as heparin and poly-Glu. Three-dimensional (3D) heteronuclear NMR experiments demonstrate that interaction with heparin reinforces the β-strand structure as well as α-helical structure in several regions of tau [60].
4 Tertiary Structure of Tau
Due to its intrinsically disordered nature, it is extremely difficult to characterize the 3D conformation of tau in the free state. Small-angle X-ray scattering studies reveal that tau forms extended conformations comparable in size with random coils [55, 65]. The average radius of gyration of 2N4R tau is about 6.5 nm from small-angle X-ray scattering measurement or 5.1 nm from single molecule Förster resonance energy transfer (FRET) measurement [65, 66]. Spin relaxation rate measurement indicates that the NTD is highly mobile, whereas the PRD and MTBD are more rigid [57]. This is consistent with single-molecule force spectroscopy characterization which shows that the MTBD is more compact than the NTD [67]. Consequently, tau monomer forms extensive dynamic intramolecular interactions which can be captured by cross-linking mass spectrometry, FRET, and paramagnetic relaxation enhancement (PRE) of NMR signals [57, 58, 61, 68, 69]. Based on the distance information from FRET signals, Jeganathan et al. proposed a paperclip-like folded model for tau monomer, where the C-terminal end of tau folds over into the vicinity of the MTBD and the N-terminal end folds onto the C-terminal end (Fig. 3a) [68]. Although the NTD is outside the FRET distance of MTBD, PRE shows that the NTD is in close contact with PRD and MTDB [57, 69]. It is noted that the paperclip-like folded conformation does not restrain tau as tau remains highly mobile throughout [68].

Tertiary and assembly structure of tau. a Schematic illustration of the paperclip-like folded conformation of tau monomer. Residues within FRET distance are indicated by arrows. b Conformational ensemble of tau monomer based on cross-linking data [71]. Five conformers are shown by different colors. c LLPS of tau under the influence of salt concentration and tau concentration. d Packing of β-sheets in the filament core of tau from AD patients (Color figure online)
To construct the atomic conformations of tau monomer, molecular modeling or simulations have been performed using restrains from PRE, residual dipolar coupling, or chemical cross-linking [57, 58, 66, 69,70,71]. The resulted conformational ensembles show that tau monomer can adopt distinct topology with variable secondary structure elements (Fig. 3b). Since different experimental techniques capture different structure information, conformational ensembles constructed using different experimental information and simulation methods may exhibit different secondary and tertiary structure properties.
Since the free energy landscape of tau conformational transition is not flat, some tau conformations may be trapped into local free energy minima. Indeed, distinct tau conformer species have been detected and isolated in experiments. Two groups of conformations with distinct degree of compactness and rotational dynamics have been identified from single molecule fluorescence anisotropy characterization using free tau molecules [72, 73]. Alternatively, two tau monomeric species are isolated by sonication treatment of tau fibrils [74, 75]. One tau form is inert and the other is seed-competent. A consensus of structural investigation on these tau species is that exposure of the PHF6* and PHF6 motifs in the MTBD is related to the formation of aggregation-prone conformations. This principle may be applicable to explain factors promoting or inhibiting tau aggregation. Pro301 mutation in the R3 region promotes tau aggregation. Recently, cross-linking mass spectrometry revealed that Pro301 mutation destabilizes local structures and extends the MTBD [61]. Phosphorylation also has remarkable influence on tau conformation. NMR derived ensembles indicate that AT8 phosphorylation expands tau [69].
5 Structure of Tau Assemblies
Tau forms a variety of assemblies, including soluble oligomers, insoluble filaments, and liquid droplets. Their structures and assembling mechanisms have been subjected to extensive studies.
5.1 Structure of Tau Oligomers
Tau forms soluble oligomers with various molecular weights. Through a sensitive split-luciferase assay, Wegmann et al. detected the formation of tau oligomers in cells and they found that stable tau dimers are released and taken up by cells [76]. Although tau oligomers are known as the major toxic species in vivo, their structures remain elusive [77, 78]. Tau contains two cysteine residues. Therefore, intermolecular disulfide bond has been found as an important factor promoting formation of tau oligomers [79,80,81,82]. However, disulfide-independent oligomerization of tau has also been observed [82,83,84], where tau monomers are held together by electrostatic interactions between the negatively charged NTD and the positively charged PRD and MTBD [85,86,87]. Furthermore, several factors can promote tau oligomerization by changing tau conformation or serving as bridging molecules [88,89,90]. The conformations of tau oligomers are heterogeneous, since some oligomers can seed monomeric tau aggregation while others cannot grow into long filaments [80, 91, 92]. Although the atomic conformations of oligomeric tau are still lacking, immunodetection suggests that the conformations of oligomeric tau are different from those of monomeric tau and aggregated tau as monoclonal antibodies raised against tau oligomers show no reactivity toward monomeric tau and tau filaments [93,94,95]. This suggestion is further supported by biophysical studies. CD characterization shows that tau oligomers contain more β-sheets than tau monomer, but the β-sheets in tau oligomers are packed differently from those in filaments [92, 96]. Bis-ANS binding experiment shows that some hydrophobic patches buried in tau monomer and fibrils are exposed in tau oligomers [96]. Molecular modeling using cross-linking restraints may be a promising strategy to construct the conformational ensembles of tau oligomers in the future.
5.2 Structure of Tau Filaments
Tau forms amyloid filaments in human brain. Different neurodegenerative diseases show distinct tau isoform compositions and filament structures [14, 97]. Many studies focus on tau filaments isolated from AD brains, indicating that the tau filament is composed of an amyloid core and a fuzzy coat. Limited proteolysis shows that the amyloid core is dominantly formed by the MTBD while the fuzzy coat consists of the N- and C-terminal domains [98,99,100]. Further spectroscopy characterization reveals that the filament core has clear cross-β structure and the fuzzy coat remains unstructured [101,102,103,104].
Tau NFTs are composed of paired helical filaments and straight filaments. Electron microscopy images show that the cross-section of tau filament core in AD has a C-shaped morphology [105, 106]. Cryo-EM structural studies reveal that paired helical filaments and straight filaments are made of two identical protofilaments comprising residues Val306–Phe378 with different inter-protofilament packing (Fig. 3d) [107, 108]. Besides AD, cryo-EM structures of filament cores from CTE, PiD and CBD have also been determined [51, 109,110,111], illustrating that different tauopathies have unique tau filament folds. R3, R4, and the N-terminal part of R’ are involved in forming the filament cores of all tauopathies studied, while R1 and R2 are exclusively present in filament cores of PiD and CBD, respectively. Unidentified fuzzy cryo-EM densities are present adjacent to the structured core, some of which turn out to be (poly)-ubiquitin chains and the P2 region [51, 112]. Heparin is widely used to induce recombinant tau aggregation. Structures of heparin-induced tau filaments have also been determined and are found different from those in diseases [113, 114]. These results suggest that cofactors and PTMs influence the structures of tau filaments [115]. However, the detailed mechanism remains unknown and has to be explored in the future.
5.3 Structure of Tau Droplet
The newest identified assembly state of tau is liquid droplet, which is formed via liquid–liquid phase separation (LLPS) (Fig. 3c). To phase separate, tau molecules undergo extensive intermolecular interactions with each other, polyanions, metal ions, or tau-associated protein [116,117,118,119,120,121,122,123,124,125,126,127]. LLPS of tau is suppressed with increased salt concentration, suggesting that electrostatic interactions are critical for the formation of tau droplets [116, 117, 119, 123]. The involvement of hydrophobic interactions in driving tau droplets formation is also observed when tau droplets are dissolved with 1,6-hexanediol [117, 128, 129]. PTMs have a marked effect on regulating the LLPS of tau. Acetylation by histone acetyltransferase p300 or CREB suppresses LLPS of tau or tau/RNA complex [43, 122]. While phosphorylation either introduced by MARK2 or SF9 insect cells promotes LLPS of tau [117, 118], mouse brain extract phosphorylated tau shows a reduced propensity of LLPS with RNA [121].
Structural characterization of tau droplets is challenging. Fusion, fission, fluorescence recovery after photobleaching and electron paramagnetic resonance spectroscopy indicate that tau droplets are in dynamic liquid state [119, 121, 122]. The level of β-structure content is increased upon LLPS [117, 123, 128]; however, the β-structure content in the droplets is still much smaller than that in the amyloid fibrils. Although the entire polypeptide chain of tau adopts more extended conformations in the droplet state [130], local conformation of the PHF6* region may remain unchanged [123].
There is mounting evidence showing that protein liquid droplets fulfill a range of biological functions and LLPS underlies the formation of membraneless compartments in living cells [131,132,133,134,135,136]. Some liquid droplets can convert into filaments or promote filaments formation, suggesting that LLPS are related to amyloid aggregation in some neurodegenerative diseases [137,138,139,140]. However, whether LLPS of tau is linked to fibril formation remains controversial. On one hand, some studies suggest that LLPS of tau mediates and facilitates aggregation. Aggregation enhancing factors, including polyanions, pro-aggregation mutations, and PTMs, promote LLPS of tau and tau droplets can turn into aggregates with elongated incubation [43, 118, 141]. On the other hand, other studies suggest that LLPS and amyloid aggregation of tau are independent processes although they occur in overlapping conditions [129]. Recently, Boyko et al. showed that tau LLPS greatly accelerates formation of fibrillar aggregates induced by heparin [124]. Since the conformations of PHF6 and PHF6* in the droplets are almost indistinguishable from those in the dilute state [123], LLPS may not accelerate tau aggregation by promoting aggregation-prone tau formation. However, the concentration of tau in the droplet is much higher than that in the dilute phase [118, 128], LLPS may promote the fibrillation reaction in a concentration-dependent regulatory mechanism [124]. Furthermore, phase separation of tau could facilitate the formation of soluble tau oligomers [142]. Consequently, although the connection between LLPS and tau fibrillation remains elusive, they may be able to influence each other.
6 Clues for Aggregation Inhibitor Design
Based on the structural knowledge of tau, aggregation inhibitors can be designed by stabilizing tau in the inert conformations or blocking the propagation of aggregation (Fig. 4). PTMs are critical factors converting tau monomers from the inert conformations to the aggregation-prone conformations. Therefore, regulating the PTMs of tau is a therapeutic approach to tauopathies. Glycogen synthase kinase 3β (GSK3β) is a major tau kinase. It phosphorylates tau at 42 sites [143]. Inhibiting the activity of GSK3β by lithium reduced the phosphorylation of tau and levels of aggregated insoluble tau [144, 145]. Novel GSK3β inhibitors include memantine and ifendropil, indirubin, BIO-acetoxime, and NP12 [146,147,148]. The acetyltransferase p300 acetylates tau a multiple sites. Salsalate treatment has been found to inhibit p300 activity and lower levels of acetylated tau [45]. However, recent studies showed that salsalate has little effect on the disease progression in PSP [149]. The major function of tau is to bind to microtubule and promote microtubule assembly. Increasing the stability of microtubule reduces tau dissociation from microtubules and the consequent aggregation. Microtubule stabilizing molecules tested includes davunetide and abeotaxane [150]. Unfortunately, clinical trials showed that both drugs are not efficient treatments for PSP or AD [151,152,153].

Strategies for aggregation inhibitor design based on tau conformational transition
Tau aggregation can be inhibited by binding to various molecules, including molecular chaperones, antibodies, and small molecules [15, 154,155,156,157,158,159,160,161,162,163,164,165]. Most of these molecules bind to the MTBD, mainly around the PHF6* and PHF6 motifs. Therefore, shielding these two motifs or interrupting their intermolecular interactions may be a general mechanism to inhibit tau aggregation. The conformational ensembles of tau monomer and the structures of tau filaments provide valuable information for tau aggregation inhibitor design/discovery. Small molecule binding sites are found by analyzing the conformational ensembles of tau K18 construct [166, 167]. Recently, Baggett and Nath identified novel tau aggregation inhibitors through a combination of molecular dynamics simulations, ensemble docking, and virtual screen of compound libraries [168]. Based on the atomic structures of amyloid fibrils formed by PHF6 and PHF6*, peptides have been designed to inhibit the aggregation of tau by capping the ends of tau fibrils [169,170,171,172,173]. Methylene blue (MB) and its derivative leuco-methylthioninium (LMTM) are efficient tau aggregation inhibitors [174]. Structural investigations suggest that MB and LMTM bind to the MTBD of tau and trap it in aggregation-incompetent conformations [32, 166]. Although LMTM effectively reduced tau pathology and improved cognition in transgenic mouse models of AD, it failed to show effects on the primary cognitive endpoints in two phase III trials [175,176,177]. Recently, it was found that while MB efficiently inhibits tau fibrils formation, it increases the number of granular tau oligomers [90]. This study provides a possible mechanistic explanation for the poor performance of LMTM in the Phase III clinical trials. Recently, Gorantla et al. designed cobalt(II)-complexes for effective inhibition of tau and disaggregation of preformed tau fibrils, illustrating potential application of metal-based therapeutics for tauopathies [178].
Antibodies can be raised to target tau fragments, monomers, oligomers, or filaments. By binding to these tau species, antibodies may inhibit the formation or propagation of aggregation-prone tau, or promote clearance of tau aggregates. Antibodies are able to cross the blood–brain barrier and reside for much longer in the body than small molecules. Consequently, immunotherapies are promising strategies for tauopathies treatment [179, 180]. Active immunization has been shown to reduce pathology of tauopathies. Two tau vaccines (i.e., AADvac1 and ACI-35) have been developed and are currently in clinical trials [181]. However, the risk of adverse immune reactions raises safety concerns on active immunization. Alternatively, the effect of passive immunization is transient and its specificity is higher. To date, several clinical trials have been conducted for various monoclonal tau antibodies in patients with AD or PSP [179].
7 Conclusions
As a major target for tauopathies treatment, tau protein has been subjected to extensive investigations. Although free tau monomer is intrinsically disordered and contains low secondary structure propensity, its conformations could be divided into two distinct classes: compacted inert conformation and extended aggregation-prone conformation. Several factors, including post-translational modifications, amino acid mutations, and interacting molecules, can modulate the conformational ensemble of tau monomer, thus inhibiting or promoting tau aggregation. Recently, 3D structures of tau filaments from several tauopathies have been determined by cryo-EM, revealing that different tauopathies have unique tau filament folds. Structural information on tau monomer and filaments provides important clues for tau aggregation inhibitor design/discovery through suppressing the formation of aggregation-prone conformations or blocking the propagation of aggregates. So far, structural knowledge on tau oligomers is very limited, although tau oligomers are toxic and can be spread between cells. Determining the 3D conformations of tau oligomers is urgent for a deeper understanding of tau aggregation and tauopathies. Novel tau aggregation inhibitors may be designed or discovered by targeting tau oligomers in the future.
References
Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2:a006247
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21
Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70:410–426
Sotiropoulos I, Galas MC, Silva JM, Skoulakis E, Wegmann S, Maina MB, Blum D, Sayas CL, Mandelkow EM, Mandelkow E, Spillantini MG, Sousa N, Avila J, Medina M, Mudher A, Buee L (2017) Atypical, non-standard functions of the microtubule associated tau protein. Acta Neuropathol Commun 5:91
Brandt R, Trushina NI, Bakota L (2020) Much more than a cytoskeletal protein: physiological and pathological functions of the non-microtubule binding region of tau. Front Neurol 11:590059
Papin S, Paganetti P (2020) Emerging evidences for an implication of the neurodegeneration-associated protein tau in cancer. Brain Sci 10:862
Dixit R, Ross JL, Goldman YE, Holzbaur EL (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319:1086–1089
Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buee L, Galas MC (2011) Nuclear tau, a key player in neuronal DNA protection. J Biol Chem 286:4566–4575
Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont J, Issad T, Gerhardt E, Pagesy P, Vileno M, Bournonville C, Hamdane M, Bantubungi K, Lancel S, Demeyer D, Eddarkaoui S, Vallez E, Vieau D, Humez S, Faivre E, Grenier-Boley B, Outeiro TF, Staels B, Amouyel P, Balschun D, Buee L, Blum D (2017) Tau deletion promotes brain insulin resistance. J Exp Med 214:2257–2269
Cruz A, Verma M, Wolozin B (2019) The pathophysiology of tau and stress granules in disease. Adv Exp Med Biol 1184:359–372
Szabo L, Eckert A, Grimm A (2020) Insights into disease-associated tau impact on mitochondria. Int J Mol Sci 21:6344
Koren SA, Galvis-Escobar S, Abisambra JF (2020) Tau-mediated dysregulation of RNA: evidence for a common molecular mechanism of toxicity in frontotemporal dementia and other tauopathies. Neurobiol Dis 141:104939
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40:189–210
Brunden KR, Ballatore C, Crowe A, Smith AB 3rd, Lee VM, Trojanowski JQ (2010) Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp Neurol 223:304–310
Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI (2012) Tau as a therapeutic target in neurodegenerative disease. Pharmacol Ther 136:8–22
Wischik CM, Harrington CR, Storey JM (2014) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88:529–539
Li C, Gotz J (2017) Tau-based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov 16:863–883
Medina M (2018) An overview on the clinical development of tau-based therapeutics. Int J Mol Sci 19:1160
Soeda Y, Takashima A (2020) New insights into drug discovery targeting tau protein. Front Mol Neurosci 13:590896
Iqbal K, Liu F, Gong CX (2016) Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 12:15–27
Andreadis A (2006) Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog Mol Subcell Biol 44:89–107
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3:519–526
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399
Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133:665–704
Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E (1994) Domains of tau protein and interactions with microtubules. Biochemistry 33:9511–9522
Chen J, Kanai Y, Cowan NJ, Hirokawa N (1992) Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360:674–677
Wegmann S, Medalsy ID, Mandelkow E, Muller DJ (2013) The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc Natl Acad Sci USA 110:E313-321
Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, Jackson S, O’Neill MJ, Isaacs AM, Hutton ML, Szekeres PG, Goedert M, Bose S (2015) Conformation determines the seeding potencies of native and recombinant tau aggregates. J Biol Chem 290:1049–1065
Chidambaram H, Chinnathambi S (2021) Role of cysteines in accelerating tau filament formation. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.07392020.01856720
Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M (2013) Mechanistic basis of phenothiazine-driven inhibition of tau aggregation. Angew Chem Int Ed Engl 52:3511–3515
Beharry C, Cohen LS, Di J, Ibrahim K, Briffa-Mirabella S, Alonso Adel C (2014) Tau-induced neurodegeneration: mechanisms and targets. Neurosci Bull 30:346–358
Almansoub H, Tang H, Wu Y, Wang DQ, Mahaman YAR, Wei N, Almansob YAM, He W, Liu D (2019) Tau abnormalities and the potential therapy in Alzheimer’s disease. J Alzheimers Dis 67:13–33
Alquezar C, Arya S, Kao AW (2020) Tau post-translational modifications: dynamic transformers of tau function, degradation, and aggregation. Front Neurol 11:595532
Hanger DP, Anderton BH, Noble W (2009) Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med 15:112–119
Kanemaru K, Takio K, Miura R, Titani K, Ihara Y (1992) Fetal-type phosphorylation of the tau in paired helical filaments. J Neurochem 58:1667–1675
Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268:24374–24384
Alonso AD, Di Clerico J, Li B, Corbo CP, Alaniz ME, Grundke-Iqbal I, Iqbal K (2010) Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem 285:30851–30860
Liu F, Gong CX (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67:953–966
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, Trojanowski JQ (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135:807–818
Ferreon JC, Jain A, Choi KJ, Tsoi PS, MacKenzie KR, Jung SY, Ferreon AC (2018) Acetylation disfavors tau phase separation. Int J Mol Sci 19:1360
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2:252
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21:1154–1162
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, Trojanowski JQ (2013) Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol 183:344–351
Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, Petrucelli L (2014) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet 23:104–116
Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L (2016) Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener 11:47
Gorsky MK, Burnouf S, Sofola-Adesakin O, Dols J, Augustin H, Weigelt CM, Gronke S, Partridge L (2017) Pseudo-acetylation of multiple sites on human tau proteins alters tau phosphorylation and microtubule binding, and ameliorates amyloid beta toxicity. Sci Rep 7:9984
Carlomagno Y, Chung DC, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, Tong J, DeTure M, Dickson DW, Petrucelli L, Cook C (2017) An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem 292:15277–15286
Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, Seyfried NT, Petrucelli L, Fitzpatrick AWP (2020) Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 180:633–644
Kametani F, Yoshida M, Matsubara T, Murayama S, Saito Y, Kawakami I, Onaya M, Tanaka H, Kakita A, Robinson AC, Mann DMA, Hasegawa M (2020) Comparison of common and disease-specific post-translational modifications of pathological tau associated with a wide range of tauopathies. Front Neurosci 14:581936
Avila J, Jimenez JS, Sayas CL, Bolos M, Zabala JC, Rivas G, Hernandez F (2016) Tau structures. Front Aging Neurosci 8:262
Wille H, Drewes G, Biernat J, Mandelkow EM, Mandelkow E (1992) Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol 118:573–584
Schweers O, Schönbrunn-Hanebeck E, Marx A, Mandelkow E (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for b-structure. J Biol Chem 269:24290–24297
Mukrasch MD, Markwick P, Biernat J, Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M (2007) Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J Am Chem Soc 129:5235–5243
Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M (2009) Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 7:e34
Schwalbe M, Ozenne V, Bibow S, Jaremko M, Jaremko L, Gajda M, Jensen MR, Biernat J, Becker S, Mandelkow E, Zweckstetter M, Blackledge M (2014) Predictive atomic resolution descriptions of intrinsically disordered hTau40 and α-synuclein in solution from NMR and small angle scattering. Structure 22:238–249
Mukrasch MD, Biernat J, von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M (2005) Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J Biol Chem 280:24978–24986
Sibille N, Sillen A, Leroy A, Wieruszeski JM, Mulloy B, Landrieu I, Lippens G (2006) Structural impact of heparin binding to full-length tau as studied by NMR spectroscopy. Biochemistry 45:12560–12572
Chen D, Drombosky KW, Hou Z, Sari L, Kashmer OM, Ryder BD, Perez VA, Woodard DR, Lin MM, Diamond MI, Joachimiak LA (2019) Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat Commun 10:2493
Karikari TK, Thomas R, Moffat KG (2020) The C291R tau variant forms different types of protofibrils. Front Mol Neurosci 13:39
Roman AY, Devred F, Byrne D, La Rocca R, Ninkina NN, Peyrot V, Tsvetkov PO (2019) Zinc induces temperature-dependent reversible self-assembly of tau. J Mol Biol 431:687–695
Ahmadi S, Wu B, Song R, Zhu S, Simpson A, Wilson DJ, Kraatz HB (2020) Exploring the interactions of iron and zinc with the microtubule binding repeats R1 and R4. J Inorg Biochem 205:110987
Mylonas E, Hascher A, Bernado P, Blackledge M, Mandelkow E, Svergun DI (2008) Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry 47:10345–10353
Nath A, Sammalkorpi M, DeWitt DC, Trexler AJ, Elbaum-Garfinkle S, O’Hern CS, Rhoades E (2012) The conformational ensembles of α-synuclein and tau: combining single-molecule FRET and simulations. Biophys J 103:1940–1949
Wegmann S, Scholer J, Bippes CA, Mandelkow E, Muller DJ (2011) Competing interactions stabilize pro- and anti-aggregant conformations of human tau. J Biol Chem 286:20512–20524
Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E (2006) Global hairpin folding of tau in solution. Biochemistry 45:2283–2293
Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M (2011) Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J Am Chem Soc 133:15842–15845
Luo Y, Ma BY, Nussinov R, Wei GH (2014) Structural insight into tau protein’s paradox of intrinsically disordered behavior, self-acetylation activity, and aggregation. J Phys Chem Lett 5:3026–3031
Popov KI, Makepeace KAT, Petrotchenko EV, Dokholyan NV, Borchers CH (2019) Insight into the structure of the “unstructured” tau protein. Structure 27:1710–1715
Manger LH, Foote AK, Wood SL, Holden MR, Heylman KD, Margittai M, Goldsmith RH (2017) Revealing conformational variants of solution-phase intrinsically disordered tau protein at the single-molecule level. Angew Chem Int Ed Engl 56:15584–15588
Foote AK, Manger LH, Holden MR, Margittai M, Goldsmith RH (2019) Time-resolved multirotational dynamics of single solution-phase tau proteins reveals details of conformational variation. Phys Chem Chem Phys 21:1863–1871
Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, Joachimiak LA, Diamond MI (2018) Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 7:e36584
Sharma AM, Thomas TL, Woodard DR, Kashmer OM, Diamond MI (2018) Tau monomer encodes strains. Elife 7:e37813
Wegmann S, Nicholls S, Takeda S, Fan Z, Hyman BT (2016) Formation, release, and internalization of stable tau oligomers in cells. J Neurochem 139:1163–1174
Ward SM, Himmelstein DS, Lancia JK, Binder LI (2012) Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans 40:667–671
Mroczko B, Groblewska M, Litman-Zawadzka A (2019) The role of protein misfolding and tau oligomers (TauOs) in Alzheimer’s disease (AD). Int J Mol Sci 20:4661
Schweers O, Mandelkow EM, Biernat J, Mandelkow E (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci USA 92:8463–8467
Kim D, Lim S, Haque MM, Ryoo N, Hong HS, Rhim H, Lee DE, Chang YT, Lee JS, Cheong E, Kim DJ, Kim YK (2015) Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci Rep 5:15231
Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E (1998) A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA 95:15712–15717
Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci 25:3020–3029
Watanabe A, Hong WK, Dohmae N, Takio K, Morishima-Kawashima M, Ihara Y (2004) Molecular aging of tau: disulfide-independent aggregation and non-enzymatic degradation in vitro and in vivo. J Neurochem 90:1302–1311
Karikari TK, Nagel DA, Grainger A, Clarke-Bland C, Hill EJ, Moffat KG (2019) Preparation of stable tau oligomers for cellular and biochemical studies. Anal Biochem 566:67–74
Rosenberg KJ, Ross JL, Feinstein HE, Feinstein SC, Israelachvili J (2008) Complementary dimerization of microtubule-associated tau protein: implications for microtubule bundling and tau-mediated pathogenesis. Proc Natl Acad Sci USA 105:7445–7450
Donhauser ZJ, Saunders JT, D’Urso DS, Garrett TA (2017) Dimerization and long-range repulsion established by both termini of the microtubule-associated protein tau. Biochemistry 56:5900–5909
Feinstein HE, Benbow SJ, LaPointe NE, Patel N, Ramachandran S, Do TD, Gaylord MR, Huskey NE, Dressler N, Korff M, Quon B, Cantrell KL, Bowers MT, Lal R, Feinstein SC (2016) Oligomerization of the microtubule-associated protein tau is mediated by its N-terminal sequences: implications for normal and pathological tau action. J Neurochem 137:939–954
Fichou Y, Oberholtzer ZR, Ngo H, Cheng CY, Keller TJ, Eschmann NA, Han S (2019) Tau-cofactor complexes as building blocks of tau fibrils. Front Neurosci 13:1339
Ramachandran G, Udgaonkar JB (2011) Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by tau protein. J Biol Chem 286:38948–38959
Soeda Y, Saito M, Maeda S, Ishida K, Nakamura A, Kojima S, Takashima A (2019) Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer’s disease. J Alzheimers Dis 68:1677–1686
Tepper K, Biernat J, Kumar S, Wegmann S, Timm T, Hubschmann S, Redecke L, Mandelkow EM, Muller DJ, Mandelkow E (2014) Oligomer formation of tau protein hyperphosphorylated in cells. J Biol Chem 289:34389–34407
Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46:3856–3861
Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI (2011) Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286:23063–23076
Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD, Dineley KT, Jackson GR, Kayed R (2014) Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34:4260–4272
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R (2012) Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 26:1946–1959
Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49:10039–10041
Goedert M, Spillantini MG (2019) Ordered assembly of tau protein and neurodegeneration. Adv Exp Med Biol 1184:3–21
Jakes R, Novak M, Davison M, Wischik CM (1991) Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J 10:2725–2729
Barghorn S, Davies P, Mandelkow E (2004) Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 43:1694–1703
Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA 85:4884–4888
Kirschner DA, Abraham C, Selkoe DJ (1986) X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-b conformation. Proc Natl Acad Sci USA 83:503–507
Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA (2003) Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-b structure. Proc Natl Acad Sci USA 100:9034–9038
Bibow S, Mukrasch MD, Chinnathambi S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M (2011) The dynamic structure of filamentous tau. Angew Chem Int Ed Engl 50:11520–11524
Sillen A, Wieruszeski JM, Leroy A, Younes AB, Landrieu I, Lippens G (2005) High-resolution magic angle spinning NMR of the neuronal tau protein integrated in Alzheimer’s-like paired helical fragments. J Am Chem Soc 127:10138–10139
Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA 88:2288–2292
Crowther RA, Wischik CM (1985) Image reconstruction of the Alzheimer paired helical filament. EMBO J 4:3661–3665
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547:185–190
Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW, Goedert M (2018) Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 136:699–708
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, Goedert M (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561:137–140
Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, Scheres SHW (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568:420–423
Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, Murayama S, Ghetti B, Hasegawa M, Goedert M, Scheres SHW (2020) Novel tau filament fold in corticobasal degeneration. Nature 580:283–287
Savastano A, Jaipuria G, Andreas L, Mandelkow E, Zweckstetter M (2020) Solid-state NMR investigation of the involvement of the P2 region in tau amyloid fibrils. Sci Rep 10:21210
Zhang W, Falcon B, Murzin AG, Fan J, Crowther RA, Goedert M, Scheres SH (2019) Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 8:e43584
Dregni AJ, Mandala VS, Wu H, Elkins MR, Wang HK, Hung I, DeGrado WF, Hong M (2019) In vitro 0N4R tau fibrils contain a monomorphic beta-sheet core enclosed by dynamically heterogeneous fuzzy coat segments. Proc Natl Acad Sci USA 116:16357–16366
Scheres SH, Zhang W, Falcon B, Goedert M (2020) Cryo-EM structures of tau filaments. Curr Opin Struct Biol 64:17–25
Hernandez-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, Alberti S, Diez S, Hyman AA (2017) Local nucleation of microtubule bundles through tubulin concentration into a condensed tau phase. Cell Rep 20:2304–2312
Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M (2017) Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein tau. Nat Commun 8:275
Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, Laskowski PR, MacKenzie D, Kamath T, Commins C, Vanderburg C, Roe AD, Fan Z, Molliex AM, Hernandez-Vega A, Muller D, Hyman AA, Mandelkow E, Taylor JP, Hyman BT (2018) Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J 37:e98049
Boyko S, Qi X, Chen TH, Surewicz K, Surewicz WK (2019) Liquid-liquid phase separation of tau protein: the crucial role of electrostatic interactions. J Biol Chem 294:11054–11059
Vega IE, Umstead A, Kanaan NM (2019) EFhd2 affects tau liquid-liquid phase separation. Front Neurosci 13:845
Lin Y, McCarty J, Rauch JN, Delaney KT, Kosik KS, Fredrickson GH, Shea JE, Han S (2019) Narrow equilibrium window for complex coacervation of tau and RNA under cellular conditions. Elife 8:e42571
Ukmar-Godec T, Hutten S, Grieshop MP, Rezaei-Ghaleh N, Cima-Omori MS, Biernat J, Mandelkow E, Soding J, Dormann D, Zweckstetter M (2019) Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat Commun 10:2909
Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, Han S (2017) RNA stores tau reversibly in complex coacervates. PLoS Biol 15:e2002183
Boyko S, Surewicz K, Surewicz WK (2020) Regulatory mechanisms of tau protein fibrillation under the conditions of liquid-liquid phase separation. Proc Natl Acad Sci USA 117:31882–31890
Rane JS, Kumari A, Panda D (2020) The acetyl mimicking mutation, K274Q in tau, enhances the metal binding affinity of tau and reduces the ability of tau to protect DNA. ACS Chem Neurosci 11:291–303
Singh V, Xu L, Boyko S, Surewicz K, Surewicz WK (2020) Zinc promotes liquid-liquid phase separation of tau protein. J Biol Chem 295:5850–5856
Wang K, Liu JQ, Zhong T, Liu XL, Zeng Y, Qiao X, Xie T, Chen Y, Gao YY, Tang B, Li J, Zhou J, Pang DW, Chen J, Chen C, Liang Y (2020) Phase separation and cytotoxicity of tau are modulated by protein disulfide isomerase and s-nitrosylation of this molecular chaperone. J Mol Biol 432:2141–2163
Ambadipudi S, Reddy JG, Biernat J, Mandelkow E, Zweckstetter M (2019) Residue-specific identification of phase separation hot spots of Alzheimer’s-related protein tau. Chem Sci 10:6503–6507
Lin Y, Fichou Y, Zeng Z, Hu NY, Han S (2020) Electrostatically driven complex coacervation and amyloid aggregation of tau are independent processes with overlapping conditions. ACS Chem Neurosci 11:615–627
Majumdar A, Dogra P, Maity S, Mukhopadhyay S (2019) Liquid-liquid phase separation is driven by large-scale conformational unwinding and fluctuations of intrinsically disordered protein molecules. J Phys Chem Lett 10:3929–3936
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18:285–298
Wu H, Fuxreiter M (2016) The Structure and dynamics of higher-order assemblies: amyloids, signalosomes, and granules. Cell 165:1055–1066
Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, Tompa P, Fuxreiter M (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol 28:420–435
Feng Z, Chen X, Wu X, Zhang M (2019) Formation of biological condensates via phase separation: characteristics, analytical methods, and physiological implications. J Biol Chem 294:14823–14835
Cramer P (2019) Organization and regulation of gene transcription. Nature 573:45–54
Rhine K, Vidaurre V, Myong S (2020) RNA droplets. Annu Rev Biophys 49:247–265
Aguzzi A, Altmeyer M (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol 26:547–558
Shin Y, Brangwynne CP (2017) Liquid phase condensation in cell physiology and disease. Science 357:eaaf4382
Alberti S, Dormann D (2019) Liquid-liquid phase separation in disease. Annu Rev Genet 53:171–194
de Oliveira GAP, Cordeiro Y, Silva JL, Vieira T (2019) Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv Protein Chem Struct Biol 118:289–331
Rane JS, Kumari A, Panda D (2019) An acetylation mimicking mutation, K274Q, in tau imparts neurotoxicity by enhancing tau aggregation and inhibiting tubulin polymerization. Biochem J 476:1401–1417
Kanaan NM, Hamel C, Grabinski T, Combs B (2020) Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun 11:2809
Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, Terro F (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev 12:289–309
Hong M, Chen DC, Klein PS, Lee VM (1997) Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 272:25326–25332
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K (2005) Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 102:6990–6995
Sereno L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sanchez MB, Gich I, Agullo JM, Perez M, Avila J, Guardia-Laguarta C, Clarimon J, Lleo A, Gomez-Isla T (2009) A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis 35:359–367
Liu X, Ou S, Yin M, Xu T, Wang T, Liu Y, Ding X, Yu X, Yuan J, Huang H, Zhang X, Tan X, Chen L, Chen Y (2017) N-methyl-D-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3β and cyclin-dependent kinase 5. Discov Med 23:221–234
Aourz N, Serruys AK, Chabwine JN, Balegamire PB, Afrikanova T, Edrada-Ebel R, Grey AI, Kamuhabwa AR, Walrave L, Esguerra CV, van Leuven F, de Witte PAM, Smolders I, Crawford AD (2019) Identification of GSK-3 as a potential therapeutic entry point for epilepsy. ACS Chem Neurosci 10:1992–2003
VandeVrede L, Dale ML, Fields S, Frank M, Hare E, Heuer HW, Keith K, Koestler M, Ljubenkov PA, McDermott D, Ohanesian N, Richards J, Rojas JC, Thijssen EH, Walsh C, Wang P, Wolf A, Quinn JF, Tsai R, Boxer AL (2020) Open-label phase 1 futility studies of salsalate and young plasma in progressive supranuclear palsy. Mov Disord Clin Pract 7:440–447
Gozes I (2011) Microtubules (tau) as an emerging therapeutic target: NAP (davunetide). Curr Pharm Des 17:3413–3417
Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, Rabinovici GD, Fagan AM, Cobigo Y, Brown JA, Jung JI, Hare E, Geldmacher DS, Natelson-Love M, McKinley EC, Luong PN, Chuu EL, Powers R, Mumford P, Wolf A, Wang P, Shamloo M, Miller BL, Roberson ED, Boxer AL (2020) Reactions to multiple ascending doses of the microtubule stabilizer TPI-287 in patients with Alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol 77:215–224
Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol JC, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Hoglinger GU, Koestler M, Jack CR Jr, Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, Investigators AL (2014) Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 13:676–685
Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M, AL-108-211 Study (2013) A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord 35:325–336
Kundel F, De S, Flagmeier P, Horrocks MH, Kjaergaard M, Shammas SL, Jackson SE, Dobson CM, Klenerman D (2018) Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high Affinity. ACS Chem Biol 13:636–646
Baughman HER, Clouser AF, Klevit RE, Nath A (2018) HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J Biol Chem 293:2687–2700
Gorantla NV, Chinnathambi S (2018) Tau protein squired by molecular chaperones during Alzheimer’s disease. J Mol Neurosci 66:356–368
McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, Goedert M, James LC (2017) Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc Natl Acad Sci USA 114:574–579
Sandusky-Beltran LA, Sigurdsson EM (2020) Tau immunotherapies: lessons learned, current status and future considerations. Neuropharmacology 175:108104
Mok SA, Condello C, Freilich R, Gillies A, Arhar T, Oroz J, Kadavath H, Julien O, Assimon VA, Rauch JN, Dunyak BM, Lee J, Tsai FTF, Wilson MR, Zweckstetter M, Dickey CA, Gestwicki JE (2018) Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat Struct Mol Biol 25:384–393
Cisek K, Cooper GL, Huseby CJ, Kuret J (2014) Structure and mechanism of action of tau aggregation inhibitors. Curr Alzheimer Res 11:918–927
Brunden KR, Trojanowski JQ, Lee VM (2009) Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov 8:783–793
Calcul L, Zhang B, Jinwal UK, Dickey CA, Baker BJ (2012) Natural products as a rich source of tau-targeting drugs for Alzheimer’s disease. Future Med Chem 4:1751–1761
Rauch JN, Olson SH, Gestwicki JE (2017) Interactions between microtubule-associated protein tau (MAPT) and small molecules. Cold Spring Harb Perspect Med 7:a024034
Pickhardt M, Neumann T, Schwizer D, Callaway K, Vendruscolo M, Schenk D, St George-Hyslop P, Mandelkow EM, Dobson CM, McConlogue L, Mandelkow E, Toth G (2015) Identification of small molecule inhibitors of tau aggregation by targeting monomeric tau as a potential therapeutic approach for tauopathies. Curr Alzheimer Res 12:814–828
Lo CH, Lim CK, Ding Z, Wickramasinghe SP, Braun AR, Ashe KH, Rhoades E, Thomas DD, Sachs JN (2019) Targeting the ensemble of heterogeneous tau oligomers in cells: a novel small molecule screening platform for tauopathies. Alzheimers Dement 15:1489–1502
Kiss R, Csizmadia G, Solti K, Keresztes A, Zhu M, Pickhardt M, Mandelkow E, Toth G (2018) Structural basis of small molecule targetability of monomeric tau protein. ACS Chem Neurosci 9:2997–3006
Chong B, Li M, Li T, Yu M, Zhang Y, Liu Z (2018) Conservation of potentially druggable cavities in intrinsically disordered proteins. ACS Omega 3:15643–15652
Baggett DW, Nath A (2018) The rational discovery of a tau aggregation inhibitor. Biochemistry 57:6099–6107
Zheng J, Liu C, Sawaya MR, Vadla B, Khan S, Woods RJ, Eisenberg D, Goux WJ, Nowick JS (2011) Macrocyclic beta-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J Am Chem Soc 133:3144–3157
Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D (2011) Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475:96–100
Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, Gonen T, Eisenberg DS (2018) Structure-based inhibitors of tau aggregation. Nat Chem 10:170–176
Seidler PM, Boyer DR, Murray KA, Yang TP, Bentzel M, Sawaya MR, Rosenberg G, Cascio D, Williams CK, Newell KL, Ghetti B, DeTure MA, Dickson DW, Vinters HV, Eisenberg DS (2019) Structure-based inhibitors halt prion-like seeding by Alzheimer’s disease-and tauopathy-derived brain tissue samples. J Biol Chem 294:16451–16464
Berhanu WM, Masunov AE (2015) Atomistic mechanism of polyphenol amyloid aggregation inhibitors: molecular dynamics study of curcumin, exifone, and myricetin interaction with the segment of tau peptide oligomer. J Biomol Struct Dyn 33:1399–1411
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA 93:11213–11218
Seripa D, Solfrizzi V, Imbimbo BP, Daniele A, Santamato A, Lozupone M, Zuliani G, Greco A, Logroscino G, Panza F (2016) Tau-directed approaches for the treatment of Alzheimer’s disease: focus on leuco-methylthioninium. Expert Rev Neurother 16:259–277
Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JM, Harrington CR, Wischik CM (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388:2873–2884
Wilcock GK, Gauthier S, Frisoni GB, Jia J, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Schelter BO, Wischik DJ, Davis CS, Staff RT, Vuksanovic V, Ahearn T, Bracoud L, Shamsi K, Marek K, Seibyl J, Riedel G, Storey JMD, Harrington CR, Wischik CM (2018) Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a phase III clinical trial. J Alzheimers Dis 61:435–457
Gorantla NV, Landge VG, Nagaraju PG, Priyadarshini Cg P, Balaraman E, Chinnathambi S (2019) Molecular cobalt(II) complexes for tau polymerization in Alzheimer’s disease. ACS Omega 4:16702–16714
Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14:399–415
Panza F, Solfrizzi V, Seripa D, Imbimbo BP, Lozupone M, Santamato A, Tortelli R, Galizia I, Prete C, Daniele A, Pilotto A, Greco A, Logroscino G (2016) Tau-based therapeutics for Alzheimer’s disease: active and passive immunotherapy. Immunotherapy 8:1119–1134
Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, Vince-Kazmerova Z, Katina S, Fialova L, Prcina M, Parrak V, Dal-Bianco P, Brunner M, Staffen W, Rainer M, Ondrus M, Ropele S, Smisek M, Sivak R, Winblad B, Novak M (2017) Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol 16:123–134
Holehouse AS, Das RK, Ahad JN, Richardson MO, Pappu RV (2017) CIDER: resources to analyze sequence-ensemble relationships of intrinsically disordered proteins. Biophys J 112:16–21
Yan R, Xu D, Yang J, Walker S, Zhang Y (2013) A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep 3:2619
Acknowledgements
This work was supported by Natural Science Foundation of Hubei Province [Grant Number 2019CFB713] and funding from Hubei University of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, D., Huang, X., Yan, L. et al. The Structure Biology of Tau and Clue for Aggregation Inhibitor Design. Protein J 40, 656–668 (2021). https://doi.org/10.1007/s10930-021-10017-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-021-10017-6