
Abstract
Tau filaments with distinct morphologies and/or isoform compositions underlie a large number of human neurodegenerative diseases. Their formation is important, because dominantly inherited mutations in MAPT, the tau gene, cause frontotemporal dementia with abundant filamentous tau inclusions. Assembly of tau may begin in a specific region of the brain, from where it spreads to other areas. It remains to be seen if the molecular species underlying tau aggregate-mediated neurodegeneration and propagation are the same or different. In the brains of mice transgenic for human mutant P301S tau, small tau filaments are the predominant seed-competent species. It has been suggested that different conformers of assembled tau may give rise to different human tauopathies, but until recently, it was not possible to study this directly. Electron cryo-microscopy can now be used to determine high-resolution structures of amyloid filaments from human brain. Paired helical and straight tau filaments of Alzheimer’s disease are ultrastructural polymorphs. Each filament core is composed of two identical protofilaments extending from G273/304-E380 (in the numbering of the 441 amino acid isoform of human tau), which adopt a combined cross-β/β-helix structure. They comprise the ends of the first or second microtubule-binding repeat (R1 or R2), the whole of R3 and R4, as well as 12 amino acids after R4. By contrast, the core of the narrow filament of Pick’s disease consists of a single protofilament extending from K254-F378 of 3R tau, which adopts a cross-β structure. It comprises the last 21 amino acids of R1, all of R3 and R4, as well as 10 amino acids after R4. Wide tau filaments of Pick’s disease, which are in the minority, consist of two narrow filaments packed against each other. The tau filament folds of Alzheimer’s and Pick’s diseases appear to be conserved between different cases of disease. These findings show that filamentous tau adopts one fold in Alzheimer’s disease and a different fold in Pick’s disease, establishing the existence of distinct conformers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Introduction
Ordered assembly of a small number of proteins into filaments characterises the majority of cases of age-related neurodegenerative diseases, including Alzheimer’s and Parkinson’s. Most cases are sporadic, but a small number is inherited in a dominant manner. Huntington’s disease is always inherited. Work carried out over the past 35 years established a causal role for filament formation in inherited forms of disease. By extrapolation, it appears likely that ordered assembly into filaments is also central for neurodegeneration in sporadic cases of disease. Tauopathies, which are characterised by the assembly of microtubule-associated protein tau, are the most common proteinopathies of the human nervous system (Table 1.1). They include Alzheimer’s disease (AD), Pick’s disease (PiD), chronic traumatic encephalopathy (CTE), tangle-only dementia, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD) and several rarer diseases.
Tau Protein
Tau monomers belong to the family of intrinsically disordered proteins that, upon ordered assembly, form structured amyloid filaments [14, 37, 85]. Their expression is largely confined to central and peripheral nerve cells, where they are highly enriched in axons [5]. However, tau assemblies are observed in both nerve and glial cells in a number of neurodegenerative diseases. Since assembly is concentration-dependent, it remains to be established if tau assemblies can form de novo in glial cells or if glial tau pathology requires the uptake of seeds from neurons.
Tau protein can be divided into an N-terminal region, a proline-rich domain consisting of two separate parts, the repeat domain and a C-terminal region. The N-terminal region projects away from the microtubule surface and is believed to interact with components of the neuronal plasma membrane. An interaction between exon 1 and annexins may help to explain the axonal localisation of tau [28], which may also be mediated, at least in part, by the axon initial segment [57]. Exon 1 of human tau contains a primate-specific sequence, which has been proposed to mediate interactions with neuronal proteins [78]. The PXXP motifs in the proline-rich region are recognised by SH3 domain-containing proteins of the Src family of non-receptor kinases, such as Fyn [56]. The repeat domain and some adjacent sequences mediate interactions between tau and microtubules. Electron cryo-microscopy (cryo-EM) has shown that each tau repeat binds to the outer microtubule surface and adopts an extended structure along protofilaments, interacting with alpha- and beta-tubulin [1, 50]. Single-molecule tracking revealed a kiss-and-hop mechanism, with a dwell time of tau on individual microtubules of about 40 ms [46, 65]. Despite these rapid dynamics, tau promotes microtubule assembly. Microtubules have stable and labile domains. Tau is most abundant in the labile domain, which has led to the suggestion that it may not stabilise microtubules, but enable them to have long labile domains [6, 71]. Less is known about the function of the C-terminal region.
Although it lacks a typical low-complexity domain, full-length tau has been reported to undergo liquid-liquid phase separation, which has been suggested to initiate aggregation and neurodegeneration [84, 92]. RNA-binding proteins may influence these processes [2]. Tau is subject to a number of post-translational modifications, including phosphorylation, acetylation, methylation, glycation, isomerisation, O-Glc-NAcylation, nitration, sumoylation, ubiquitination and truncation [37].
Tau Isoforms
Six tau isoforms ranging from 352 to 441 amino acids in length are expressed in adult human brain from a single MAPT gene (Fig. 1.1a) [31]. They differ by the presence or absence of inserts of 29 or 58 amino acids (encoded by exons 2 and 3, with exon 3 being only transcribed in conjunction with exon 2) in the N-terminal half and inclusion, or not, of the 31 amino acid microtubule-binding repeat, encoded by exon 10, in the C-terminal half. Inclusion of exon 10 results in the production of three isoforms with four repeats (4R) and its exclusion in a further three isoforms with three repeats (3R). The repeats comprise residues 244–368, in the numbering of the 441 amino acid isoform. In adult human brain, similar levels of 3R and 4R tau are expressed [32]. The finding that a correct 3R and 4R tau isoform ratio is essential for preventing neurodegeneration came as a surprise. 2 N isoforms are underrepresented in comparison with isoforms that include exon 2 or exclude exons 2 and 3; 2 N, 1 N and 0 N isoforms make up 9%, 54% and 37%, respectively. Big tau, which carries an additional large exon in the N-terminal half, is only expressed in the peripheral nervous system [20, 34].

Human brain tau isoforms and disease-causing MAPT mutations
(a) MAPT and the six tau isoforms expressed in adult human brain. MAPT consists of 14 exons (E). Alternative mRNA splicing of E2 (red), E3 (green) and E10 (yellow) gives rise to six tau isoforms (352–441 amino acids). The constitutively spliced exons (E1, E4, E5, E7, E9, E11, E12 and E13) are shown in blue. E6 and E8 (violet) are not transcribed in human brain. E4a (orange) is only expressed in the peripheral nervous system. The repeats (R1-R4) are shown, with three isoforms having four repeats (4R) and three isoforms with three repeats (3R). The core sequences of tau filaments from Alzheimer’s disease (G273/305-E380) determined by cryo-EM are underlined (in orange); the core sequences of tau filaments from Pick’s disease (K254-F378 of 3R tau) are also underlined (in grey). (b) Mutations in MAPT in FTDP-17T. Fifty coding region mutations and ten intronic mutations flanking E10 are shown
The expression of isoforms is not conserved. Thus, in adult mouse brain, 4R tau is exlusively present, whereas adult chicken brain expresses 3R, 4R and 5R isoforms [89]. One hyperphosphorylated 3R tau isoform lacking N-terminal repeats is characteristic of developing vertebrates. In mice, the switch from 3R to 4R tau occurs between postnatal days 9 and 18, with tau phosphorylation decreasing over time [81]. However, isoform switching and phosphorylation are regulated differently. Adult 4R isoforms are better at promoting microtubule assembly than the 3R isoform expressed during development, which is also more phosphorylated than both 3R and 4R tau in adult brain [32]. This is consistent with the need for a more dynamic cytoskeleton during the development of nerve cells.
Tau Assemblies
Brain tau can assemble into filamentous inclusions [7, 37]. The repeats and some adjoining sequences form the filament core, with the N-terminal half and the C-terminus giving rise to the fuzzy coat [30, 87, 88]. Tau filaments from brain, and those assembled in vitro from expressed protein, have a cross-β structure characteristic of amyloids [4]. Since the region that binds to microtubules also forms the core of tau filaments, physiological function and pathological assembly may be mutually exclusive.
Phosphorylation negatively regulates the ability of tau to interact with microtubules and filamentous tau is abnormally hyperphosphorylated [44]. However, it remains to be proved that phosphorylation is the trigger for tau assembly in human diseases. Alternatively, a change in conformation as part of assembly may lead to hyperphosphorylation. Since tau is hydrophilic, it is not surprising that unmodified and full-length protein requires cofactors, such as sulphated glycosaminoglycans, nucleic acids or fatty acids, to assemble into filaments [35, 48, 68, 86]. Cofactors other than heparin and/or post-translational modifications may cause the assembly of tau in human brain [25, 26].
Besides phosphorylation, other post-translational modifications may also play a role. Early studies on tau acetylation reported that it can promote both phosphorylation and assembly [15, 59]. However, subsequent work suggested an inverse correlation between acetylation and phosphorylation, with acetylation inhibiting tau assembly [11, 17]. These discrepancies may have resulted from the use of enzymes that acetylated different residues. Site-specific acetylation of K280 has been shown to enhance heparin-induced tau aggregation in vitro, while reducing microtubule assembly [40]. Unlike phosphorylation, acetylation occurs on lysine residues.
In AD, CTE, tangle-only dementia and other tauopathies, all six tau isoforms are present in disease filaments (Table 1.2). Pick bodies are made of only 3R tau. In PSP, CBD, AGD and other diseases, isoforms with 4R tau are found in the filaments. The morphologies of tau filaments vary in different diseases, even when they are mainly made of the same isoforms.
Genetics of MAPT
The relevance of tau inclusion formation for neurodegeneration became clear in June 1998, when dominantly inherited mutations in MAPT were shown to cause a form of frontotemporal dementia that can be associated with parkinsonism (FTDP-17T, also known as familial FTLD-tau) [43, 70, 76]. Abundant filamentous tau inclusions are present in either nerve cells or in both nerve and glial cells. Aβ deposits, a defining feature of AD, are not characteristic of FTDP-17T. This work established that a pathological pathway leading from monomeric to assembled tau is sufficient for causing neurodegeneration and dementia.
Sixty mutations in MAPT have been identified in FTDP-17T (Fig. 1.1b). Filaments are composed of either 3R or 4R tau, or of both 3R and 4R tau. MAPT mutations account for approximately 5% of cases of FTLD and are concentrated in exons 9–12 (encoding R1-R4) and the introns flanking exon 10. They can be divided into those with a primary effect at the protein level and those affecting the alternative splicing of tau pre-mRNA. There is no obvious correlation between known mutations and post-translational modifications of tau.
Mutations that act at the protein level change or delete single amino acids, reducing the ability of tau to interact with microtubules [41]. Some mutations also promote the assembly of tau into filaments [36, 63]. Mutations with a primary effect at the RNA level are intronic or exonic and increase the alternative mRNA splicing of exon 10. This affects the ratio of 3R to 4R isoforms, resulting in the relative overproduction of 4R tau and its assembly into filaments [43, 76]. One mutation (ΔK280) has been reported to cause the relative overexpression of 3R tau and its assembly into filaments [82].
Assembled tau shows different isoform patterns and filament morphologies, depending on the mutations in MAPT [29]. Mutations V337 M in exon 12 and R406W in exon 13 give rise to insoluble tau bands of 60, 64 and 68 kDa and a weaker band of 72 kDa. Following dephosphorylation, six bands are present that align with recombinant tau, like what is seen in AD [33]. By electron microscopy, paired helical filaments (PHFs) and straight filaments (SFs) are present. The brains of many individuals with missense MAPT mutations in exons 9–13 (K257T, L266V, S305N, G272V, L315R, S320F, S320Y, P332S, Q336H, Q336R, K369I, E372G and G389R) are characterised by abundant Pick bodies made predominantly of 3R tau. As in sporadic PiD, insoluble tau shows strong bands of 60 and 64 kDa. However, variable amounts of 68- and 72 kDa bands are also present. A third pattern is characteristic of MAPT mutations that affect the alternative mRNA splicing of exon 10, resulting in the relative overproduction of 4R tau (intronic mutations and exonic mutations N279K, L284L, L284R, ΔN296, N296D, N296H, N296N, S305L, S205N and S305S). Insoluble tau runs as two strong bands of 64 and 68 kDa, and a weaker band of 72 kDa; following dephosphorylation, three bands are present that align with recombinant 4R tau (isoforms of 383, 412 and 441 amino acids). A similar pattern of pathological tau bands is observed for mutations in exon 10, such as P301L and P301S, which have their primary effects at the protein level. Assembly of 4R tau has also been described for mutations I260V in exon 9, K317N in exon 11, E342V in exon 12 and N410H in exon 13, showing that it is possible to alter 3R and 4R tau mRNAs through mutations located outside exon 10.
The effects of MAPT mutations can vary. Neighbouring mutations in exon 12 (G335S, G335V, Q336H, Q336R and V337M) give rise to structurally distinct assemblies and exert different functional effects. Mutation G335S is characterized by abundant filamentous tau inclusions in nerve cells and glial cells, in the absence of Pick bodies [77]. Mutations Q336H and Q336R give rise to what is essentially a familial form of PiD, with abundant Pick bodies in nerve cells [69, 80], whereas mutation V337M produces a neuronal filamentous tau pathology like that of AD [70, 75]. These findings on MAPT mutations in three adjacent codons reinforce the view that the mechanisms underlying the formation of neurofibrillary lesions and Pick bodies are closely related. Recombinant tau with the G335S, G335V [64], or V337 M mutation shows a greatly reduced ability to promote microtubule assembly. By contrast, mutations Q336H and Q336R increase the ability of tau to promote microtubule assembly. Mutations G335V and V337M fail to increase heparin-induced assembly into filaments significantly, whereas mutations Q336H and Q336R increase the assembly of 3R, but not 4R, tau.
The architecture of MAPT on chromosome 17q21.31 is characterized by two haplotypes as the result of a 900 kb inversion (H1) or non-inversion (H2) polymorphism [79]. Inheritance of the H1 haplotype of MAPT is a risk factor for PSP, CBD, Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS), but not for PiD [3, 16, 22, 61, 66, 90]. The association with PD and ALS is particularly surprising, since they are not characterized by tau inclusions.
Based on genome-wide association studies for PSP and CBD, it has been shown that association with an allele at the MOBP/SLC25A38 locus results in elevated levels of appoptosin, a protein that activates caspase-3, which can cleave tau [93]. This may cause aggregation of 4R tau. Additional loci were unique to PSP or CBD. Association of the H1 haplotype with PSP had a higher odds ratio than that between apolipoprotein E epsilon 4 (APOEε4) and AD [42]. APOEε4 is the major risk factor allele for late-onset AD [19]. H1 expresses more exon 10-containing mRNA than H2, especially in subcortical regions [9, 54]. Moreover, H2 is associated with increased expression of exon 3 of MAPT in grey matter, suggesting that inclusion of exon 3 may protect against PSP, CBD, PD and ALS [10]. In experimental studies, exon 3-containing tau isoforms (those with both N-terminal inserts) have been found to assemble less than those lacking this exon [94]. Even though all six tau isoforms give rise to PHFs and SFs, known mutations in MAPT do not cause AD. They give rise to FTLD-tau. Tau with an A152T substitution has been reported to be a risk factor for AD [18], as well as for PSP, CBD and unusual tauopathies [18, 49, 53, but see also 67].
Heterozygous microdeletions of chromosome 17q21.31 give rise to a multisystem disorder with intellectual disability, hypotonia and distinct facial features (17q21.31 microdeletion syndrome or Koolen-de Vries syndrome) [51, 73, 74]. In addition to MAPT, three protein-coding genes (CRHR1, SPPL2C and KANSL1) and two putative genes (MGC57346 and CRHR-ITI) are found in this region. Deletions arise on the H2 haplotype through low-copy, repeat-mediated, nonallelic homologous recombination.
The 17q21.31 microdeletion syndrome is caused by haploinsufficiency of KANSL1, which encodes a chromatin modifier that influences gene expression through the acetylation of lysine 16 of histone H4 [52, 95]. A 50% reduction in tau levels does therefore not appear to have a detrimental effect on development of the human brain.
Neurodegeneration and Propagation
Disease-causing mutations in MAPT have made it possible to produce transgenic rodent lines that form abundant tau filaments and exhibit neurodegeneration (Figs. 1.2 and 1.3) [37]. Tau assembly correlates with neurodegeneration. Reducing assembly and increasing aggregate degradation are therefore therapeutic objectives. Since assembly is concentration-dependent, decreasing the level of soluble, monomeric tau is likely to result in reduced assembly [21, 55]. However, the molecular species of assembled tau that are responsible for neurodegeneration remain to be identified [58]. At a cellular level, it has been reported that the removal of senescent brain cells leads to a reduction in both tau assembly and neurodegeneration in transgenic mice [8]. In tauopathies, as in most neurodegenerative diseases, inclusion formation manifests many years before clinical symptoms. In future, it will therefore be important to identify individuals at risk of disease. Early diagnostics,in particular imaging of tau inclusions, is therefore likely to play an important role [38].

Electron microscopy and immunoelectron microscopy of nerve cells in brain and spinal cord from mice transgenic for human mutant P301S tau
(a, b), Cerebral cortex; (c, d), Brainstem; (e, f), Spinal cord. (b, d, f), Higher magnifications of parts of the cytoplasmic regions from (a, c, e). The electron micrographs in (c, d) show immunogold labelling of filaments using the phosphorylation-dependent anti-tau antibody AT8. Scale bars in (C) and (E) must be in micrometers: C, 1.5 m; E, 5.5 m (for a, e); F, 300 nm (for b, d, f)

Nerve cell loss in spinal cord of mice transgenic for human mutant P301S tau
(a, b), Haematoxylin and eosin-stained sections of the ventral grey matter of the lumbar spinal cord (L2-L3) from 6-month-old non-transgenic (a) and transgenic (b) mice. Swollen, abnormal material-containing motor neurons are indicated by arrows. Arrowheads point to atrophic motor neurons, with dashed arrows pointing to pyknotic cells that are surrounded by glial cells, suggestive of neuronophagia. (c) Graph showing the density of motor neurons (expressed as number of neurons per millimetre square) in the anterior horn of the lumbar spinal cord of 6-month-old non-transgenic (Control) and transgenic (P301S) mice. Nerve cell numbers were determined in five consecutive sections from each animal, with the density of motor neurons from each section being represented by a circle. The results are expressed as the means ± S.E.M. (n = 3). ∗p < 0.0001. Scale bar in (B) must be in micrometers: 60 m (for a, b)
Transgenic mouse lines were also essential for the identification of the prion-like properties of assembled tau in vivo (Fig. 1.4) [62]. Aggregation of hyperphosphorylated tau was induced following intracerebral injection of tau seeds from mice transgenic for human mutant 0N4R P301S tau into transgenic mice expressing wild-type non-aggregated 2N4R tau and, to a lesser extent, following intracerebral injection into wild-type mice [12]. Tauopathy then spread to connected brain regions, indicative of seed endocytosis, seeded aggregation, intracellular transport and release of tau seeds. This work was complemented by studies, which showed that short tau filaments have the greatest seeding activity (Figs. 1.5 and 1.6) [45]. Seeded aggregation of tau was dependent on the ability of expressed, monomeric tau to aggregate [23].

Induction of filamentous tau pathology in mice transgenic for 2N4R wild-type human tau (line ALZ17) following injection with brain extract from symptomatic mice transgenic for 0N4R P301S tau
Staining of the hippocampal CA3 region of 18-month-old ALZ17 mice with anti-tau antibodies AT8 and AT100 and Gallyas-Braak silver. Non-injected (left), 15 months after injection with brain extract from non-transgenic control mice (middle) and 15 months after injection with brain extract from 6-month-old mice transgenic for human P301S tau (right). The sections were counterstained with haematoxylin. The Scale bar must be given in micrometers

Seeding of tau assembly with sucrose gradient fractions from the brains of mice transgenic for human mutant 0N4R P301S tau in a cell-based assay
The mice were aged 4.4 weeks (no symptoms, no tau filaments) or 24.4 weeks (symptoms, abundant tau filaments). Sucrose gradient fractions were used to seed assembly of tau in HEK cells expressing 1N4R P301S tau. The pellet from a 1000,000 g spin of the seeded cells was analysed by immunoblotting for total tau (antibody DA9) and tau phosphorylated at S202/T205 (antibody AT8). Filamentous tau runs at 68 kDa (high molecular weight, HMW) and non-filamentous tau at 59 kDa (low molecular weight, LMW). As a positive control, sarkosyl-insoluble tau extracted from unfractionated brains of symptomatic transgenic P301S tau mice was used for seeding; the normalized positive control consisted in seeding with sarkosyl-insoluble tau that had been normalised for total tau levels relative to those of the sucrose gradient fractions. Seeding correlated with the presence of the 64 kDa band in 24.4-week-old mice (20–50% sucrose gradient fractions). No seeding was observed upon addition of sucrose gradient fractions from 4.4-week-old mice

Tau species in the 40% sucrose gradient fractions from the brains of symptomatic mice transgenic for human mutant P301S tau (24.4 weeks) mediate seeding and spreading in brain
(a) Unilateral injection of brainstem lysate (positive control) from symptomatic transgenic mice into the hippocampus of asymptomatic transgenic mice led to the accumulation and spread of pathology (detected by antibody PG-5, which recognises tau phosphorylated at S409) in the injected (∨) and contralateral (∧) hippocampus. Mice injected with brainstem lysate of non-transgenic mice (negative control) had minimal pathology. Injection of the 40% sucrose gradient fraction also showed substantial accumulation and spread of pathology (40% fraction), whereas mice injected with the 10% sucrose gradient fraction (10% fraction) were indistinguishable from those injected with brainstem lysate from non-transgenic mice. Injection of brainstem lysate [positive control (normalised)] that had been normalised for total tau did not show robust tau aggregate propagation. (b) In the ipsilateral hippocampus, PG-5 immunoreactivity was highest for the positive control, followed by the 40% sucrose fraction. No significant increase in signal was observed for the normalised positive control or the 10% fraction (relative to the negative control). (c) In the contralateral hippocampus, a similar, but overall milder, pattern of tau pathology was observed compared to the injecteded side. The Scale bar must be given in micrometers. ∗p < 0.05, ∗∗p < 0.01, ♯p < 0.01 for all groupwise comparisons
Distinct conformers of assembled tau appear to exist, reminiscent of prion strains. They may explain the variety of human tauopathies. Inclusions formed and spread of pathology occurred after intracerebral injection of brain homogenates from cases of AD, tangle-only dementia, PSP, CBD and AGD into a mouse line transgenic for wild-type 4R tau [13]. PiD, the filamentous inclusions of which consist of only 3R tau, was an exception. Inclusions formed at the injection sites, but spreading was not observed. However, PiD is only rarely a pure 3R tauopathy, since it can be associated with AD-type tau pathology. We therefore cannot exclude that the activity in the PiD homogenate, which induced aggregation at the injection site, may have been due to the presence of a small amount of aggregated 4R tau.
Sequence requirements for seeded tau aggregation in vivo remain to be defined. Tau assemblies reminiscent of those in the corresponding human diseases were observed following the injection of brain homogenates from patients with PSP, CBD and AGD, which are 4R tauopathies [13]. Although these findings are consistent with the existence of distinct tau aggregate conformers, the definition of such conformers must be structural.
High-Resolution Structures of Tau Filaments from Alzheimer’s Disease
By cryo-EM, high-resolution structures of Tau filaments were obtained from the frontal cortex of four individuals with AD, three sporadic and one inherited (Fig. 1.7) [25, 27]. The cores of tau filaments are made of two protofilaments consisting of residues G273/304-E380, which adopt a combined cross-β/β-helix structure (Fig. 1.8). Murine and human tau are identical in sequence in this region. The N-terminal part of the cross-β structure includes hexapeptide 306VQIVYK311, which is essential for the oligomerisation of recombinant tau and its assembly into filaments [72, 83]. It packs against 373THKLTF378, in agreement with the predicted heterozipper interaction between 306VQIVYK311 and 375KLTFR379 [60]. Constructs K18 and K19 end at E372 [39]; they can therefore not give rise to the human brain tau folds determined thus far.

Cryo-EM structures of paired helical filaments (PHFs) and straight filaments (SFs) from the frontal cortex of four AD cases. All structures show identical pairs of C-shaped protofilaments and the same inter-protofilament packing in PHFs and SFs. Cases 1, 2 and 3 had sporadic AD, whereas case 4 had inherited AD (mutation V717F in the amyloid precursor protein gene). All cases had a majority of PHFs and a minority of SFs. Yellow arrows point to the extra densities, which were present in PHFs and SFs from all four cases, bordering the solvent-exposed side-chains of R349 and K375, and of H362 and K369

Cryo-EM densities and atomic models of paired helical filaments (PHFs) and straight filaments (SFs) from the frontal cortex of one AD case
PHFs and SFs were resolved to 3.2 Å and 3.3 Å, respectively. Sharpened, high-resolution maps are shown in blue (PHFs) and green (SFs). Unsharpened 4.5 Å low-pass filtered densities are shown in grey. The models comprise G273-E380 of 3R tau and G304-E380 of 4R tau. The protofilaments of PHFs and SFs are identical, but differ in inter-protofilament packing (ultrastructural polymorphs)
Each protofilament is made of eight β-strands, five of which give rise to two regions of antiparallel β-sheets, with the other three forming a β-helix (Fig. 1.9). The C-terminal residues of R1 and R2 form part of the first β-strand. R3 contributes three and R4 four β-strands, with the final β-strand being formed by 12 amino acids after R4 (residues K369-E380). Strands β1 and β2 pack against β8, β3 packs against β7, with β4, β5 and β6 giving rise to the C-shaped β-helix.

Schematic view of the tau protofilament core of AD. The observed eight β-strand regions (β1-β8) are shown as arrows. Each filament consists of two protofilaments
PHFs and SFs are made of identical protofilaments, but differ in inter-protofilament packing, showing that they are ultrastructural polymorphs. PHF protofilaments are arranged base-to-base and SF protofilaments back-to-base. In PHFs, protofilaments are stabilised by backbone hydrogen bonds between their 332PGGGQ336 sequences. Moreover, the side-chains of K331 from one protofilament project towards the side-chains of Q336 and E338 of the other protofilament, suggesting additional interactions that stabilise the protofilament interface. Furthermore, in the protofilament interface of the PHF, extra densities between the side-chains of K331 of one protofilament and the backbone of V337 of the other have been observed. They may correspond to a solvent molecule or a post-translational modification of K331, such as mono-methylation [25].
In SFs, the protofilaments pack asymmetrically. Their backbones are nearest each other between residues 321KCGS324 of the first and 313VDLSK317 of the second protofilament. The inter-protofilament packing appears to be stabilised through the region of additional density that interacts with the side-chains of K317, T319 and K321 of both protofilaments. This density may correspond to residues 7EFE9, which constitute the N-terminal region of the discontinuous epitope of conformational anti-tau antibodies ALZ-50 and MC-1 (the C-terminal epitope is 313VDLSKVTSKC322) [47]. A similar density also interacts with K317, T319 and K321 in PHFs, where it does not contribute to the protofilament interface.
High-Resolution Structures of Tau Filaments from Pick’s Disease
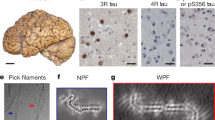
By cryo-EM, high-resolution structures of Tau filaments were determined from the frontotemporal cortex of an individual with PiD [24]. Two types of filament could be distinguished: a majority of narrow Pick filaments (NPFs) and a minority of wide Pick filaments (WPFs) (Fig. 1.10). The core of NPFs is made of a single protofilament that consists of residues K254-F378 of 3R tau, which adopt an elongated cross-β structure. Murine and human tau are identical in sequence in this region, with the exception of residue 257 (K in human, R in mouse tau). WPFs are formed by the association of two NPF protofilaments at their distal tips, where they form tight contacts through van der Waals interactions. Each protofilament comprises nine β-strands, which are arranged into four cross-β packing stacks and are connected by turns and arcs (Fig. 1.11). R1 provides two β-strands and R3 and R4 three β-strands each. These stacks pack together in a hairpin-like fashion: β1 against β8, β2 against β7, β3 against β6 and β4 against β5. The final strand, β9, is formed from the ten amino acids after R4 and packs against the opposite side of β8.

(a) Sharpened, high-resolution cryo-EM map of narrow Pick filaments with the atomic model of the Pick fold overlaid (3.2 Å resolution). Narrow (93%) and wide (7%) Pick filaments are characteristic of PiD. Unsharpened cryo-EM densities of a narrow (b) and wide (c) Pick filament. Wide Pick filaments comprise two narrow filaments that are joined at their distal tips

Schematic view of the tau protofilament core of PiD. The observed nine β-strands (β1-β9) are shown as arrows. Narrow Pick filaments are made of one and wide Pick filaments of two protofilaments
Three regions of less well-resolved density bordering the solvent-exposed faces of β4, β5 and β9 are apparent in both NPFs and WPFs. They may represent less ordered, heterogeneous and/or transiently occupied structures. The density bordering β4 is similarly located, but more extended, than that found to interact with the side-chains of K317, T319 and K321 in AD filaments.
It was not previously known why only 3R tau is present in Pick body filaments and why S262 is not phosphorylated. Our results suggest that despite sequence homology, the structure formed by K254-K274 of R1 is inaccessible to the corresponding residues from R2 (S275-S305). Moreover, because of steric constraints, the filament structure precludes phosphorylation of S262.
Conclusion
Biochemistry, molecular biology and human genetics have shown that the ordered assembly of tau is at the heart of a large number of neurodegenerative diseases, including AD, tangle-only dementia, CTE, PiD, PSP, CBD and AGD. They may be different clinical diseases, because of the existence of distinct conformers of assembled tau. Assembly leads to propagation of pathology and neurodegeneration, which are characteristic of all tauopathies. It remains to be seen if the same or different molecular species of assembled tau account for propagation and neurodegeneration. Small filaments are the major species of assembled tau responsible for the propagation of pathology.
Cryo-EM of filaments from human brain has established that distinct conformers of aggregated tau are characteristic of AD and PiD [91].Even though both types of filament share residues G273-F378 of 3R tau, their structures are very different (Fig. 1.12). Whereas PHFs and SFs of AD are made of two identical C-shaped protofilaments that each comprises eight β-strands and a combined β-sheet/β-helix structure, NPFs of PiD are made of a single elongated protofilament comprising nine β-strands and stacks of β-sheets. WPFs consist of two NPFs joined through their distal tips. Cryo-EM studies of tau filaments from additional sporadic and inherited cases of AD, as well as negative-stain immunoelectron microscopy of tau filaments from multiple cases of AD and PiD, indicated that the cores of tau filaments from each disease case contain the same sequences. It therefore seems that the structures of tau filaments are distinct between diseases, but identical in different individuals with AD or PiD. It appears likely that additional folds of assembled tau remain to be discovered in other tauopathies.

Comparison of Alzheimer and Pick tau filament folds depicted as single rungs
Paired helical and straight tau filaments of AD consist of two identical C-shaped protofilaments that differ in inter-protofilament packing (ultrastructural polymorphs). More than 90% of tau filaments of PiD are narrow filaments that consist of a single J-shaped protofilament. Wide Pick filaments consist of two narrow filaments packed against each other. C322 (yellow) and D348 (red) are highlighted. C322 is on the inside in the Alzheimer fold and the outside in the Pick fold, whereas D348 is on the outside in the Alzheimer fold and the inside in the Pick fold
References
Al-Bassam J, Ozer RS, Safer D, Halpain DS, Milligan RA. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol. 2002;157:1187–96.
Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, Ferragud A, Botelho E, Balance HI, Dhawan U, et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 2018;21:72–80.
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–5.
Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc Natl Acad Sci U S A. 2003;100:9034–8.
Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–8.
Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci. 1996;16:3601–19.
Brion JP, Passareiro H, Nunez J, Flament-Durand J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch Biol. 1985;95:229–35.
Bussian TJ, Aziz A, Meyer CF, Swenson BL, Van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–82.
Caffrey TM, Joachim C, Paracchini S, Esiri MM, Wade-Martins R. Haplotype-specific expression of exon 10 at the human MAPT locus. Hum Mol Genet. 2006;15:3529–37.
Caffrey TM, Joachim C, Wade-Martins R. Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol Aging. 2008;29:1923–9.
Carlomagno Y, Chung D, Yue M, Castanedes-Casey M, Madden BJ, Dunmore J, Tong J, DeTure M, Dickson DW, Petrucelli L, et al. An acetylation-phosphorylation switch that regulates tau aggregation propensity and function. J Biol Chem. 2017;292:15277–86.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40.
Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–47.
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VMY. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:52.
Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, Wiederholt W, Hansen L, Masliah E, Thal LJ, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997;41:277–81.
Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, et al. Acetylation of the KXGS motifs in tau is a critical determinant ion modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23:104–16.
Coppola G, Chinnathambi S, Lee JJ, Dombrowski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21:3500–12.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late-onset families. Science. 1993;261:921–3.
Couchie D, Mavilia C, Georgieff IS, Liem RK, Shelanski ML, Nunez J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:4378–81.
De Vos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481.
Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montag P, Munoz DG, et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol. 2000;47:374–7.
Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, Jackson S, O’Neill MJ, Isaacs AM, Hutton ML, et al. Conformation determines the seeding potencies of native and recombinant tau aggregates. J Biol Chem. 2015;290:1049–65.
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, Goedert M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561:137–40.
Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW, Goedert M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018;136:699–708.
Fichou Y, Lin Y, Rauch JN, Vigers M, Zeng Z, Srivasta M, Keller TJ, Freed JH, Kosik KS, Han S. Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc Natl Acad Sci U S A. 2018;115:13234–2239.
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–90.
Gauthier-Kemper A, Suárez-Alonso M, Sündermann F, Niewidok B, Fernandez MP, Bakota L, Heinisch JJ, Brandt R. Annexins A2 and A6 interact with the extreme N-terminus of tau and thereby contribute to tau’s axonal localization. J Biol Chem. 2018;293:8065–76.
Ghetti B, Oblak LA, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46.
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–5.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26.
Goedert M, Jakes R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–30.
Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–68.
Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:1183–9.
Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3.
Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–11.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40:189–210.
Goedert M, Yamaguchi Y, Mishra SK, Higuchi M, Sahara N. Tau filaments and the development of positron emission tomography tracers. Front Neurol. 2018;9:70.
Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–22.
Haj-Yahya M, Lashuel HA. Protein semisynthesis provides access to tau disease-associated post-translational modifications (PTMs) and paves the way to deciphering the tau PTM code in health and diseased states. J Am Chem Soc. 2018;140:6611–21.
Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–10.
Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12:15–27.
Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, Landi A, Szekeres PG, Murray TK, Ahmed Z, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J Neurosci. 2016;36:762–72.
Janning D, Igaev M, Sündermann F, Brühmann J, Beutel O, Heinisch JJ, Bakota L, Piehler J, Junge W, Brandt R. Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell. 2014;25:3541–51.
Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–32.
Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–9.
Kara E, Ling H, Pittman AM, Shaw K, de Silva R, Simone R, Holton JL, Warren JD, Rohrer JD, Xiromerisiou G, et al. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol Aging. 2012;33(2231):e7–e14.
Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E. Near-atomic model of microtubule-tau interactions. Science. 2018;360:1242–6.
Koolen DA, Viussers LE, Pfundt R, De Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001.
Koolen DA, Kramer JM, Neveling K, Nilldsen WM, Moore-Barton HL, Elmslie EV, Toutain A, Amiel J, Malan V, Tsai AC, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44:639–41.
Kovacs GG, Wöhrer A, Ströbel T, Botond G, Attems J, Budka H. Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol. 2011;30:3–10.
Kwok JBJ, Teber ET, Loy C, Hallupp M, Nicholson G, Mellick GD, Buchanan DD, Silburn PA, Schofield PR. Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann Neurol. 2004;55:329–34.
Lasagna-Reeves CA, de Haro M, Hao S, Park J, Rousseaux MWC, Al-Ramahi I, Jafar-Nejad P, Vilanova-Velez L, See L, De Maio A, et al. Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron. 2016;92:407–18.
Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–77.
Li X, Kumar Y, Zempel H, Mandelkow EM, Biernat J, Mandelkow E. Novel diffusion barrier for axonal retention of tau in neurons and its failure in neurodegeneration. EMBO J. 2011;30:4825–37.
Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci Res. 2006;54:197–201.
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66.
Moore CL, Huang MH, Robbenolt SA, Voss KR, Combs B, Gamblin TC, Goux WJ. Secondary nucleating sequences affect kinetics and thermodynamics of tau aggregation. Biochemistry. 2011;50:10876–86.
Morris HR, Baker M, Yasojima K, Houlden H, Khan MN, Wood NW, Hardy J, Grossman M, Trojanowski JQ, Revesz T, et al. Analysis of tau haplotypes in Pick’s disease. Neurology. 2002;59:443–5.
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM, Mandelkow EM, Mandelkow E, Buée L, Goedert M, et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun. 2017;5:99.
Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–9.
Neumann M, Diekmann S, Bertsch U, Vanmassenhove B, Bogerts B, Kretzschmar HA. Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics. 2005;6:91–5.
Niewidok B, Igaev M, Sündermann F, Janning DF, Bakota L, Brandt R. Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau’s interaction with microtubules in axon-like processes. Mol Biol Cell. 2016;27:3537–49.
Pastor P, Ezquerra M, Munoz E, Marti MJ, Blersa R, Tolosa E, Olivas R. Significant association between the tau gene A0/A0 genotype and Parkinson’s disease. Ann Neurol. 2000;47:242–5.
Pastor P, Moreno F, Clarimón J, Ruiz A, Combarros O, Calero M, López de Munain A, Bullido MJ, de Pancorbo MM, Carro E, et al. MAPT H1 haplotype is associated with late-onset Alzheimer’s disease risk in APOEɛ4 noncarriers: Results from the dementia genetics Spanish consortium. J Alzheimers Dis. 2016;49:343–52.
Pérez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–90.
Pickering-Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, Simpson SA, Moore JW, Snowden JS, de Silva R, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127:1415–26.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25.
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW. Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol. 2018;28:2181–9.
Sawaya MR, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7.
Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. Discovery of previously identified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–42.
Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, et al. Microdeletion encompassing MAPT at chromosome 17q21.31 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–7.
Spillantini MG, Crowther RA, Goedert M. Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol. 1996;92:42–8.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–41.
Spina S, Murrell JR, Yoshida H, Ghetti B, Bermingham N, Sweeney B, Dlouhy SR, Crowther RA, Goedert M, Keohane C. The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol. 2007;113:461–70.
Stefanoska K, Volkerling A, Bertz J, Poljak A, Ke YD, Ittner LM. An N-terminal motif unique to primate tau enables differential protein-protein interactions. J Biol Chem. 2018;293:3710–9.
Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37.
Tacik P, DeTure M, Hinkle KM, Lin WL, Sanchez-Contreras M, Carlomagno Y, Pedraza O, Rademakers R, Ross OA, Wszolek ZK, et al. A novel tau mutation in exon 12, p.Q336H, causes hereditary Pick disease. J Neuropathol Exp Neurol. 2015;74:1042–52.
Tuerde D, Kimura T, Miyasaka T, Furusawa K, Shimozawa A, Hasegawa M, Ando K, Hisanaga SI. Isoform-independent and –dependent phosphorylation of microtubule-associated protein tau in mouse brain during postnatal development. J Biol Chem. 2018;293:1781–93.
Van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P. The ΔK280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25.
Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β-structure. Proc Natl Acad Sci U S A. 2000;97:5129–34.
Wegmann S, Eftakharzadeh B, Tepper K, Zoltowaka KM, Bennett RE, Dujardin S, Laskowski PR, MacKenzie D, Kamath T, Commins C, et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018;37:e98049.
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–62.
Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. Am J Pathol. 1997;150:2181–95.
Wischik CM, Novak M, Thøgersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4506–10.
Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4884–8.
Yoshida H, Goedert M. Molecular cloning and functional characterization of chicken brain tau: Isoforms with up to five tandem repeats. Biochemistry. 2002;41:15203–11.
Zhang CC, Zhu JX, Wan Y, Tan L, Wang HF, Yu JT, Tan L. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. 2017;8:4494–45007.
Zhang W, Falcon B, Murzin AG, Fan J, Crowther RA, Goedert M, Scheres SHW. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife. 2019;8:43584. Biorxiv
Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, Han S. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017;15:e2002183.
Zhao Y, Tseng IC, Heyser CH, Rockenstein E, Mante M, Adame A, Zheng Q, Huang T, Wang X, Arslan PE, et al. Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron. 2015;87:963–75.
Zhong Q, Congdon EE, Nagaraja HN, Kuret J. Tau isoform composition influences rate and extent of filament formation. J Biol Chem. 2012;287:20711–9.
Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, Stefanini C, Mercuri E, Chiurazzi P, Neri G, Marangi G. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44:636–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Goedert, M., Spillantini, M.G. (2019). Ordered Assembly of Tau Protein and Neurodegeneration. In: Takashima, A., Wolozin, B., Buee, L. (eds) Tau Biology. Advances in Experimental Medicine and Biology, vol 1184. Springer, Singapore. https://doi.org/10.1007/978-981-32-9358-8_1
Download citation
DOI: https://doi.org/10.1007/978-981-32-9358-8_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9357-1
Online ISBN: 978-981-32-9358-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)