
Abstract
Sym004 is an equimolar mixture of two monoclonal antibodies, futuximab and modotuximab, which non-competitively block the epidermal growth factor receptor (EGFR). Sym004 has been clinically tested for treatment of solid tumors. The present work characterizes the non-linear pharmacokinetics (PK) of Sym004 and its constituent antibodies and investigates two types of covariate models for interpreting the interindividual variability of Sym004 exposure. Sym004 serum concentration data from 330 cancer patients participating in four Phase 1 and 2 trials (n = 247 metastatic colorectal cancer, n = 87 various types advanced solid tumors) were pooled for non-linear mixed effects modeling. Dose regimens of 0.4–18 mg/kg Sym004 dosed by i.v. infusion weekly or every 2nd week were explored. The PK profiles for futuximab and modotuximab were parallel, and the parameter values for their population PK models were similar. The PK of Sym004 using the sum of the serum concentrations of futuximab and modotuximab was well captured by a 2-compartment model with parallel linear and saturable, Michaelis–Menten-type elimination. The full covariate model including all plausible covariates included in a single step showed no impact on Sym004 exposure of age, Asian race, renal and hepatic function, tumor type and previous anti-EGFR treatments. The reduced covariate model contained statistically and potentially clinically significant influences of body weight, albumin, sex and baseline tumor size. Population PK modeling and covariate analysis of Sym004 were feasible using the sum of the serum concentrations of the two constituent antibodies. Full and reduced covariate models provided insights into which covariates may be clinically relevant for dose modifications and thus may need further exploration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enhanced activity of the epidermal growth factor receptor (EGFR) plays a key role in malignant tumor growth in solid tumors via overexpression, gene amplification, enhanced ligand production and/or mutations. Blocking EGFR has proven to provide a clinically meaningful anti-tumor activity in various solid tumors [1]. Approved monoclonal antibodies (mAbs) targeting EGFR include cetuximab, panitumumab and necitumumab. The anti-tumor activity of these agents is, however, moderate [2] and acquired resistance develops in virtually all patients over time [3]. Therefore, novel strategies are being developed to improve the efficacy of anti-EGFR therapies. One promising approach for this is Sym004 which is a mixture of two anti-EGFR antibodies, futuximab and modotuximab. The Sym004 mixture has demonstrated synergy in preclinical studies [4,5,6], and has been tested in Phase 1 and 2 clinical studies for treatment of metastatic colorectal cancer (mCRC) and other solid tumors.
The two constituent antibodies of Sym004 are manufactured as individual drug substances before being mixed at a 1:1 ratio into the Sym004 drug product (DP) (Fig. 1a). The two mouse-human chimeric IgG1 monoclonal antibodies bind to non-overlapping epitopes on the EGFR in a non-competitive manner and are thus able to bind simultaneously to their target [5]. The dual blocking induces rapid internalization and degradation of the EGFR in in vitro and in vivo preclinical studies (Fig. 1b) and this contributes to Sym004’s enhanced anti-tumor activity as compared to that of single anti-EGFR mAbs. Sym004 is dosed as the mixture and separate ELISA methods are used for quantifying serum concentrations of futuximab and modotuximab in post-dose samples (Fig. 1c). Brief descriptions of Sym004 human PK based on non-compartmental (NCA) methods have been reported previously as part of trial-specific reports [7,8,9]. In keeping with the pharmacokinetics (PK) of other anti-EGFR antibodies, the PK of Sym004 has been shown to be non-linear [7] which can be attributed due to the large abundance of EGFR in normal tissues, and hence pronounced elimination via target mediated drug disposition (TMDD) [10].

Concepts of Sym004 mixture manufacturing, mode of action and bioanalysis. a The two antibodies are manufactured as separate drug substances (DS) and mixed in a 1:1 ratio in the drug product (DP). b Futuximab and modotuximab are binding to non-overlapping epitopes on EGFR and their binding induce internalization and degradation of EGFR from the cell surface, c in a post-dose clinical serum sample, the concentrations [ ] of futuximab and modotuximab are assessed in separate ELISAs, and their sum derived as the Sym004 serum concentration
In this paper, we report the first population pharmacokinetic model for two antibodies binding independently to the same target using a TMDD-type model. Usually, separate PK models are used for compounds dosed as a mixture, but we here demonstrate that a TMDD model based on the sum of the serum concentration of the two constituent antibodies is a viable approach. Finally, an exploratory covariate analysis was performed, which estimated the effects on Sym004 PK of patient characteristics in both a full covariate model with all potentially relevant covariates, and a reduced model where only statistically significant and potentially clinically relevant covariate effects were included. Both full and reduced covariate models are important for drug development and regulatory purposes.
Methods
Clinical studies
Clinical PK data from 330 patients participating in four Sym004 Phase 1 and 2 trials (described elsewhere [7,8,9, 11]) were pooled for the population PK analysis. Dosing regimens and PK sampling schemes are summarized in Table 1. Briefly, Study 1 was a combined dose escalation and dose expansion study in advanced solid tumors and mCRC [7], Study 2 was a Phase 2 trial in advanced recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) [8], Study 3 was a randomized, double-blind Phase 2b trial in mCRC [11], and Study 4 was a combined Phase 1 dose escalation and dose expansion study in Japanese subjects with advanced solid tumors or esophageal squamous cell cancer (ESCC) [9]). In earlier reports, futuximab and modotuximab were termed mAb 992 and mAb 1024, respectively.
A wide dose range of 0.4 to 18 mg/kg was included in the trials. Sym004 was dosed by i.v. infusion every week (Q1W) or every 2nd week (Q2W), or as a 9 mg/kg loading dose followed by 6 mg/kg weekly (9/6 mg/kg weekly).
Studies included in the analysis were conducted in accordance with Good Clinical Practice guidelines and the ethical principles that have their origin in the Declaration of Helsinki. The protocols and informed consent forms were approved by the institutional review boards or ethics committees and participants provided written informed consent before any study-related procedures were performed.
Bioanalysis and immunogenicity
Serum concentrations of futuximab and modotuximab were determined using anti-idiotypic bivalent antibody fragments (F[ab’]2) specific for either futuximab or modotuximab in two separate competitive ELISA formats (Fig. 1c), with a validated range for each of the antibodies from 0.5 µg/mL to 20 µg/mL. More details, including method for determination of anti-drug binding antibodies (ADA) can be found in Supplementary Materials App. 1.
Population pharmacokinetic analyses
Software and model selection criteria
The population PK model and covariate analyses for Sym004 were performed using non-linear mixed effects modeling NONMEM software (version 7.3.0, ICON Development Solutions Ellicott City, MD, USA) [12]. The data and modeling results were processed using Microsoft R Open (MRO) Version 3.4.4 software [13]. Simulations were performed using NONMEM, Perl Speaks NONMEM (PsN) Version 4.4.8 [14], and the RxODE package in R [15].
The model parameters were estimated using the first-order conditional estimation method with interaction (FOCE-I). The confidence intervals (CI) of the estimates in the covariate models were calculated using four different methods:
- (a)
default RSR sandwich estimator in NONMEM,
- (b)
the cross-product matrix S estimator in NONMEM,
- (c)
the sampling importance re-sampling method (SIR) [16],
- (d)
bootstrap [17] using 500 data sets sampled with replacement.
The variance–covariance matrix M used in the default sandwich estimator in NONMEM is given by:
where the R matrix is the Hessian of 2ln(likelihood) with respect to the model parameters and the S matrix is the cross-product matrix [18].
Discrimination between models was made based on changes in the objective function value and inspection of graphical diagnostics. Standard continuous-data goodness of fit (GOF) plots were used for the assessment of model adequacy for the population PK models. Visual predictive checks (VPCs) [19] were additionally used in model assessment, and to determine whether the observed population profiles could be appropriately simulated using the population PK model.
Model development
Exploratory graphical analysis suggested that the PK of Sym004 (and its constituent antibodies) is non-linear and that a second distribution compartment exists. Hence, a 2-compartment model with both a linear elimination and a saturable, Michaelis–Menten (MM) type elimination was used as the initial model. The structure and the parameterization are shown schematically in Fig. 2, and the differential equations for the model are shown in Eqs. 1 and 2. A similar model structure was used for modeling the PK of futuximab and modotuximab as separate entities.
where A1 is the amount in the central compartment, A2 is the amount in the peripheral compartment, k12= Q/V1, and k21= Q/V2.

Structure of the Sym004 base PK model. CL linear clearance, V1 central volume of distribution, V2 peripheral volume of distribution, Q intercompartmental clearance, CLMM clearance via Michaelis–Menten elimination, Vmax maximum velocity for MM elimination, Km Sym004 serum concentration for 50% of Vmax, C serum concentration of Sym004
A combined additive plus proportional model was used to describe the residual variability (Eq. 3).
where Cij is the observed drug concentration at the ith individual at time j, Ĉij is the corresponding model-predicted drug concentration, and ε1,ij and ε2,ij represent the proportional and additive residual random errors, respectively.
Exponential random effect models (parameterized as additive on log-scale) were used to describe the IIV for the structural model parameters in the population PK model (Eq. 4).
where \(\theta_{i,k}\) is the estimate for the kth parameter for the ith subject, θk is the typical population estimate of the kth parameter, and ηi,k is the parameter for the individual deviation from θk.
Parameters for IIV were introduced and retained in the model, provided estimates were neither very small nor inclusion caused instability in the model fitting procedure. Two types of correlation between the IIVs were also used during model development: (a) diagonal—assuming the IIVs to independent and (b) a full correlation matrix which estimates the correlation between all the IIVs in the model.
Covariate model building
A full covariate model for Sym004 was built by incorporating all prespecified potential covariate relationships into the base model in a single step—the so-called “full model approach” [20]. Prior to building the full covariate model, potential correlations between the covariates were evaluated graphically. If two or more covariates were highly correlated (taken as correlation > 0.6), then the most clinically/biologically and practically relevant covariate was tested in the analysis.
Table 2 provides an overview of which continuous and categorical covariates were tested on which PK parameters and specifies if covariates were provided as their baseline value or whether they were time-varying covariates; tumor size, defined as the sum of diameters of the target lesions according to the guideline for assessing tumor burden (Recist 1.1. [21]), was included as both a baseline and time-varying covariate.
A summary of the baseline covariates across all four studies is provided in Supplementary Materials App. 2. The covariate effects for continuous covariates were scaled so that the effect was quantified as a fold-change across 95% of the covariate range:
where θ1 is the population value of the estimated PK parameter, PARi the individual-specific realization for the ith subject with the value of the covariate (COVi) scaled to a combination of the median covariate value (COV) and 97.5th and 2.5th quantiles of the covariate (Q97.5 and Q2.5). We believe this is a novel approach and is an extension of the commonly used method of estimating covariate effects scaled to the median value of the covariate.
Taking the exponent of the θ2 estimate and its 95% confidence interval, results in the estimated fold-change in PK parameter due to the covariate across 95% of the covariate range. Categorical covariates were similarly estimated as shift on the logarithmic scale and back-transformed to a fold-change. By scaling to the covariate range, the size of the estimated fold-change may be used to identify appropriately large covariate effects in the full covariate model.
Body weight at baseline used as the measure of body size, was considered a structural component of the model and was retained in the population PK model throughout covariate model building.
The reduced covariate model, containing only statistically, and sufficiently large covariates, was derived from the full covariate model so that only covariates with effect size point estimates outside the range of 0.80 to 1.25, and with 95% confidence intervals not overlapping 1 were retained. Covariate effects and their confidence intervals were graphically represented using Forest plots [22] for both the full and reduced covariate models.
Simulations
The reduced covariate model was used to simulate the average steady state concentrations (Css). PK parameters were sampled from the estimated parameter distributions, and either a loading dose of 9 mg/kg, followed by 10 weekly doses of 6 mg/kg, or 11 weekly flat doses of 400 mg were applied. All observed albumin baseline concentrations (n = 330) were combined with a range of body weights from 35 to 125 kg in 5 kg steps. The resultant 6270 covariate combinations were combined with sampled eta values and used to simulate the profiles underlying the Css calculations. The median and the area encompassing 90% of the simulated subjects for each weight category were plotted by weight.
Results
Bioanalysis and immunogenicity
A total number of 5341 Sym004 PK samples from the four studies from 330 patients were included, after excluding 7% of the data points associated with protocol violations or with an absolute weighted residual (WRES) larger than 4. Re-inclusion of the WRES-outliers in the final model did not impact magnitude of parameter estimates, just their uncertainty. Sym004 was not detectable in any baseline samples and the in-study validation showed successful incurred sample reanalysis results (at least two-thirds (67%) of the incurred sample reanalysis results were within 30% of the original sample results). A confirmed positive treatment induced anti-Sym004 ADA response was detectable in 2/336 patients (0.6%). For both patients, ADA became detectable with low titers (< 80) in samples taken in the follow-up period when Sym004 was cleared from the circulation.
Exploratory PK data analysis and base model
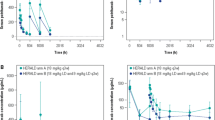
The PK profiles of futuximab and modotuximab were found to be approximately parallel (Fig. 3), suggesting that the distribution and elimination of the two antibodies proceeds in a similar fashion following dosing in the 1:1 ratio in the Sym004 drug product. The terminal half-life of the two antibodies and hence of Sym004 increased with increasing dose, suggesting the presence of pronounced non-linearity. The PK profile shape also suggested the presence of a second distributional compartment. Hence, the 2-compartment model with a MM-type saturable elimination was applied for the base model of Sym004. The base model structure provided a good fit to the data and population parameter estimates were estimated with good precision (Supplementary Materials App. 3). The proportional residual error was low (17.6%); the additive residual error was 1.68 µg/mL which is reasonably close to the bioanalytical assay LLOQ (0.5–1.0 µg/mL, Supplementary Materials App. 1). The IIVs for the four parameters V1, V2, CL and Vmax were in the range of 18–39% with CL having the largest IIV (Table 3). The VPC across all doses demonstrated a good description of the data and its variability, and the GOF plots showed no trends with study, dose, time or predicted Sym004 concentration (Supplementary Materials App. 3 and 6). VPCs stratified by dose similarly showed no dose level-specific model misfit (results not shown). The base model was found to be adequate for simulating PK profiles of Sym004 based on each patient’s dosing history as shown in Fig. 4.

Serum concentrations of futuximab, modotuximab and Sym004 versus time after the most recent dose (for time points from 2 weeks and onwards). Selected dose level shown for clarity. Grey lines and black dots: individual profiles for Sym004. Solid thick lines: median profiles, Blue: Sym004, orange: modotuximab, red: futuximab (Color figure online)

Example of Sym004 concentration versus time profiles. Example concentration profiles for mCRC patients, loading dose of 9 mg/kg followed by weekly doses of 6 mg/kg, and dosing interruptions. Blue line: individual predicted curve, red markers: observed Sym004 concentrations, the horizontal black line is the LLOQ for Sym004 (Color figure online)
The simulations in Fig. 5 using the base model typical parameters illustrate that the amount eliminated via the MM elimination route is saturated for weekly dosing of 6 mg/kg and higher. The MM pathway contributed 25–50% of the total amount of Sym004 being eliminated for dose levels of 6–12 mg/kg where the MM was saturated (Fig. 5).

Simulations of elimination due to MM elimination of Sym004. Percentage of total elimination of Sym004 due to MM elimination for weekly dosing of 1.5–12 mg/kg Sym004. Simulations performed in R using the typical parameters from the base PK model
The Vmax and Km estimates for futuximab and modotuximab were each about 50% of the Sym004 values, whereas the remaining PK parameters were similar for the two antibodies and for Sym004 (Supplementary Materials App. 4). Thus, the approach of using the sum of futuximab and modotuximab for modeling of Sym004 PK was supported. Since futuximab and modotuximab are both full IgG antibodies, they have identical molecular weights (150 kDa) and hence it is feasible to sum their µg/mL concentrations rather than their molar concentrations.
Covariate analysis—development of the full covariate model
The baseline covariates across the four studies are summarized in Supplementary Materials App. 2. The covariates included in the covariate scope (Table 2) were added to the base model, after initial removal of three non-informative covariates with very small effect sizes (< 0.6% change in parameter). Furthermore, the IIV structure was simplified to Vmax, CL and V1 in a diagonal matrix in order to obtain SE estimates. The Forest plot for the fold change of the covariates included in the full covariate model is presented in Fig. 6, including a comparison of the four methods for CI estimation. All the covariate effects were reasonably precisely estimated using the sandwich estimator (Supplementary Materials App. 5). The condition number was 1017, which suggests that the full model was over-parameterized.

Forest plot for the full covariate model. Vertical dotted lines indicate the limits associated with 0.8 to 1.25-fold parameter change across 95% of the covariate range for continuous covariates, and versus reference for categorical covariates. SIR sample importance re-sampling
The Forest plot (Fig. 6) illustrates which covariates might warrant dose modifications (effect size outside 0.80–1.25-fold), which have no clinical relevance (effect size within 0.80–1.25-fold), useful for regulatory claims of no effect, and for which more information may be needed (CIs wide or overlapping limits) [23]. The largest covariate influence was for body weight on Vmax, CL, V1 and V2. The majority of the covariates were associated with effect size point estimates within the 0.80–1.25 range. Of note, the effects of Asian race (primarily Japanese) on CL and Vmax were insignificant as were prior treatment with bevacizumab, ECOG status, age, and tumor type. Inclusion of the covariates in the full model resulted in a substantial reduction in the unexplained variability for Vmax and CL, and a modest reduction for V1 (Table 3).
Covariate analysis—development of the reduced model
The reduced covariate model was derived from the full covariate model by retaining only covariates with effect size point estimates outside the range of 0.80 to 1.25, and with 95% confidence intervals not overlapping 1. As shown in the Forest plot in Fig. 7 and in Table 4, the only covariates fulfilling these criteria were for weight on V1, V2, CL and Vmax, an association of higher CL with low albumin, a 21% lower CL for females, and a higher Vmax for higher baseline tumor size. The precision of the estimates was good; the reduced covariate model had a condition number of 53, which indicates that the over-parameterization seen in the full covariate model was effectively negated. The CIs for the four methods were overlapping, indicating that the estimated CIs are robust in the reduced covariate model. Both CL and Vmax varied approximately 1.7-fold between body weights of 50 and 90 kg (Fig. 8). Smaller effects were seen for the influence of albumin on CL and tumor size on Vmax (Fig. 8). Similar reductions in the estimated IIVs were obtained as with the full covariate model (Table 3), indicating that the most important covariates for explaining the IIVs were retained in the reduced model.

Forest plot for the reduced covariate model. Vertical dotted lines indicate the limits associated with 0.8 to 1.25-fold parameter change across 95% of the covariate range for continuous covariates, and versus reference for categorical covariates. SIR sample importance re-sampling

Key covariate relationships in the reduced covariate model. Red and blue circles are individual estimates for females and males, respectively; the orange solid line is a loess smooth (Color figure online)
Equations 6–9 outline the calculation of the model parameters for the individual subject i in the reduced covariate model:
where sex = 1 for females and sex = 0 for males.
Discussion
A Michaelis–Menten approximation [25] of the full TMDD model [24] was used to capture the non-linear PK of Sym004. The model performed well over the large range of dose levels and serum concentrations included in the analysis. The PK of other anti-EGFR antibodies have been modelled using a similar model structure [26, 27]. The linear clearance for Sym004 of 14.1 mL/h is somewhat higher than for endogenous IgG antibodies and fully human antibodies with linear PK (10.5 mL/h, [28]), but this is commonly seen for chimeric antibodies [29]. The central and peripheral volumes of distribution were 3.5 L and 2.4 L, respectively, and were thus within the range expected for monoclonal antibodies [28]. The MM pathway was demonstrated to contribute substantially to the total Sym004 elimination also for dose regimens saturating the TMDD (Fig. 5). This is consistent with the large amount of EGFR in the body.
For the models of futuximab and modotuximab, the values for Vmax and Km were each about 50% of the values for Sym004. This is as expected since they contribute equally to the Sym004 concentration in the Sym004 model. It also in accordance with the full TMDD model [24] when assuming that the binding affinity and rate of internalization after binding to the EGFR of the two antibodies are similar: the Km parameter is a combination of the Kd (on/off rates) for target binding and kint, the rate of internalization of the complex between the target and the mAb [25]; Vmax is the product of total amount of target (Rtot) and the kint [25]. In vitro studies indicate that the Kd values for binding to human EGFR are approximately 50 pM for both antibodies [5] whereas they act synergistically on EGFR internalization [5]. The synergistic effect on internalization would theoretically result in a larger Vmax (due to larger kint) for the mixture than for the individual antibodies. The phenomenon was supported by data in non-human primates [6] where the half-life of futuximab and modotuximab were considerably longer when they were dosed alone as compared to dosing them together in the Sym004 mixture. It is, however, not possible to deduce this for the clinical setting, where the antibodies have only been dosed together as the Sym004 mixture. Most investigations of full TMDD models for drug combinations have focused on drugs binding competitively to the same targets [30, 31], whereas the TMDD model for drug–drug interaction for two drugs binding non-competitively to the same target as described by Koch et al. [32] is what comes closest to the mechanisms underlying the Sym004 PK. This mechanistic TMDD model was deemed not to be feasible for fitting the Sym004 PK data as estimation of all parameters would necessitate data from dosing of the antibodies as individual entities. The simple MM approximation provided a good description of the data and accurate estimation of parameters although it did not provide insight into the mechanisms for the PK interactions of the two antibodies.
A full covariate modeling approach rather than a stepwise p value inclusion/reduction of the model was used to allow for an unbiased evaluation of prespecified covariate relationships [23, 33]. Despite initial removal of highly correlated covariates, the full covariate model may still suffer from over-parameterization, and the choice of SE estimation method could impact the assessment of covariate importance. The CIs calculated by the four methods were comparable for well-defined covariate effects. Bootstrap CIs were slightly wider than for the sandwich method, whereas SIR CI estimates—that are reported to have strong theoretical advantages [16]—tended to provide the narrowest CIs. For covariate effects which were poorly estimated (e.g. tumor type, Asian race), the computationally simpler option of MATRIX = S provided overly inflated estimates for the SE. Hence, for full covariate models, which are frequently over-parameterized and present computational difficulties in practice, care should be taken choosing the method for calculation of SE, and MATRIX = S should be avoided.
The reduced covariate model was obtained by, in a single step, retaining only covariates with effect size point estimates outside the range of 0.80 to 1.25, and with 95% confidence intervals not overlapping 1. Different approaches could have been taken to derive the reduced model from the full model, and different approaches could have resulted in potentially different reduced models. By presenting both the full and reduced models the influence of covariates not present in the reduced model but present in the full model is clear and independent of the approach taken to obtain the reduced model. The reduced covariate model for Sym004 contained statistically significant and sufficiently large influences of weight, albumin, sex and baseline tumor size. The positive association of V1, V2, CL and Vmax with body weight is a common finding for monoclonal antibodies [34], including cetuximab [35] and panitumumab [27]. A 1.7-fold difference in Sym004 clearance for low versus high body weight was observed. Thus, the interpatient variability in exposure is lower for the currently applied body weight-based dosing as compared to using a hypothetical, fixed (flat) mg dose per patient (Fig. 9). However, this does not rule out that a flat dosing scheme could be employed if it can be shown (via PK/PD modelling) that the higher PK variability associated with flat dosing would not affect safety and efficacy responses.

Simulation of the average serum concentration of Sym004 in steady state (Css). a Weight-based dosing of 9/6 mg kg and b flat dosing of 400 mg (both weekly dosing). Css = steady state AUC after the 11th dose divided by the dose interval. Red (females) and blue (males) circles: observed empirical Bayes estimates for the 330 patients. Orange line: simulated median. Blue area 90% CI for the simulations. Simulations using the reduced covariate model and sampling from eta distributions of PK parameters (Color figure online)
As has been observed for other antibodies [34], low levels of albumin were associated with a higher clearance of Sym004, corresponding to a 35% difference across 95% of the observed albumin range. Hypoalbuminemia is a well-recognized marker for elevated protein turnover frequently observed in cancer [34]. The elevated protein turnover explains the higher clearance of exogenous proteins such as Sym004. Azzopardi et al. [35] also reported an association between low baseline albumin and high clearance for cetuximab. Whether low albumin levels should call for increasing the dose of Sym004 is unclear since the inherent poor prognosis associated with hypoalbuminemia may not be mitigated by increasing the exposure to cancer therapeutics as was shown recently for trastuzumab [36].
The association of an increased Vmax in patients with a large baseline tumor burden may be explained by either the contribution to TMDD from the EGFR in the tumor, or, an underlying association of a large tumor size with poor patient health as in the case for albumin. Glassman and Balthasar [37] showed in a PBPK model, that a tumor of 20 mL (corresponding to a diameter of 34 mm if assuming a spherical shape) forms only 0.012% of the total EGFR in the body. Although EGFR is frequently overexpressed in tumor tissue the contribution from EGFR in the tumor to the Vmax is likely to be modest. The modest contribution is supported by the observation that the PK of the anti-EGFR mAb panitumumab was not associated with the expression levels of EGFR in the tumor [27]. For the anti-HER2 antibody trastuzumab, the number of metastatic sites was associated with a higher clearance although the overall effect on exposure was not clinically relevant [38].
Sym004 CL in females was 21% lower as compared to males even after compensating for the difference in body weight. The difference in PK parameters and exposure between males and females is also illustrated in Figs. 8 and 9. The difference is not expected to warrant dose adjustments, unless female patients are proven to have more serious side effects than males and that may be related to higher exposure.
The reduced covariate model allows simulations of potentially clinically relevant covariate effects to support clinical trial design, and enables preselection of covariates for a confirmatory covariate analyses [39]. The standard criteria for bioequivalence of a 0.80–1.25-fold difference for clinical relevance were used to evaluate the statistical importance of the covariates. These criteria are frequently applied in the absence of exposure–response analyses that define the therapeutic window. The future development of a PK/PD model may potentially explain the counter-intuitive dose–response for Sym004 observed in mCRC where the 9/6 mg/kg weekly dosing resulted in a more favorable overall survival than the higher dose level of 12 mg/kg weekly [11]. It is hypothesized that this may be caused by the more frequent dose interruptions and dose reductions due to adverse events such as skin rash observed for the 12 mg/kg group. The future development of PK/PD models for both efficacy and safety will allow the clinical relevance of all PK covariate effects to be appropriately determined, along with the need (or not) for dose adjustments.
Conclusion
The PK profiles of the two antibodies were parallel despite futuximab and modotuximab being two different antibodies, indicating that underlying PK properties like clearance, TMDD and volume of distribution are similar. This is the first time a population PK model for a mixture of two antibodies binding to non-overlapping sites on the same target has been reported. It is unique that the PK of Sym004 can be described by the sum of the concentration of the two antibodies instead of requiring a model for each antibody separately. The full and reduced covariate models provide a robust basis for evaluating potential dose modification of Sym004.
References
Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti A, Batra SK (2012) Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets 16:15–31
You B, Chen EX (2012) Anti-EGFR monoclonal antibodies for treatment of colorectal cancers: development of cetuximab and panitumumab. J Clin Pharmacol 52:128–155
Van Emburgh BO, Andrea Sartore-Bianchic A, Di Nicolantonioa F, Siena S, Bardelli AA (2012) Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol Oncol 8:1084–1094
Koefoed K, Stenaa L, Søderberg JN, Kjær I, Jacobsen HJ, Meijer P-J, Haurum JS, Jensen A, Kragh M, Andersen PS, Pedersen MW (2011) Rational identification of an optimal antibody mixture for targeting the epidermal growth factor receptor. MAbs 3:584–595
Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, Kragh M (2010) Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res 70:588–597
Skartved NJ, Jacobsen HJ, Pedersen MW, Jensen PF, Jørgensen TK, Hey A, Kragh M (2011) Preclinical pharmacokinetics and safety of Sym004: a synergistic antibody mixture directed against epidermal growth factor receptor. Clin Cancer Res 7:5962–5972
Dienstmann R, Patnaik A, Garcia-Carbonero R, Cervantes A, Benavent M, Rosello S, Tops BB, van der Post RS, Argiles G, Skartved NJ, Hansen UH, Hald R, Pedersen MW, Kragh M, Horak ID, Braun S, Van Cutsem E, Tolcher AW, Tabernero J (2015) Safety and activity of the first-in-class Sym004 anti-EGFR antibody mixture in patients with refractory colorectal cancer. Cancer Discov 5:1–12
Machiels JP, Specenier P, Krauss J, Dietz A, Kaminsky MC, Lalami Y, Henke M, Keilhotz U, Knecht R, Skartved NJ, Horak ID, Pamperin P, Braun S, Gauler TC (2015) A proof of concept trial of the anti-EFGR antibody mixture Sym004 in patients with squamous cell carcinoma of the head and neck. Canc Chemother Pharmacol 76:13–20
Kojima T, Yamazaki K, Kato K, Muro K, Hara H, Chin K, Goddemeier T, Kuffel S, Watanabe M, Doi R (2018) Phase I dose-escalation trial of Sym004, an anti-EGFR antibody mixture, in Japanese patients with advanced solid tumors. Cancer Sci 109:3253–3262
Levy G (1994) Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther 56:248–252
Montagut C, Argiles G, Ciardiello F, Poulsen TT, Dienstmann R, Kragh M, Kopetz S, Lindsted T, Ding C, Vidal J, Clausell-Tormos J, Siravegna G, Sanchez-Martin FJ, Koefoed K, Pedersen MW, Grandal MM, Dvorkin M, Wyrwicz L, Rovira A, Cubillo A, Salazar R, Desseigne F, Nadal C, Albanell J, Zagonel V, Siena S, Fumi G, Rospo G, Nadler P, Horak ID, Bardelli A, Tabernero J (2018) Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses: a phase 2 randomized clinical trial. JAMA Oncol 4:e175245
Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ (eds) (1989–2013) NONMEM users guides. Icon Development Solutions, Ellicott City
R Development Core Team (2016). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. http://www.R-project.org
Lindbom L, Pihlgren P, Jonsson EN (2005) PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79:241–257
Wang Q, Hallow KM, James DA (2016) A Tutorial on RxODE: simulating differential equation pharmacometric models in R. CPT Pharmacomet Syst Pharmacol 5:3–10
Dosne AG, Bergstrand M, Karlsson MO (2017) An automated sampling importance resampling procedure for estimating parameter uncertainty. J Pharmacokinet Pharmacodyn 44:509–520
Efron B, Tibshirani RJ (1994) An introduction to the bootstrap. CRC Press/Chapman & Hall, Boca Raton
Aoki Y, Nordgren R, Hooker AC (2016) Preconditioning of nonlinear mixed effects models for stabilisation of variance–covariance matrix computations. AAPS J 18:505–518
Karlsson MO, Holford N (2008) A tutorial on visual predictive checks. PAGE 2008. Abstr 1434 [www.page-meeting.org/?abstract=1434]
Gastonguay M (2011) Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies. PAGE 2011 [www.page-meeting.org/?abstract=2229]
Eisenhauer EA, Therasse P, Bogaerts H, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij JJ (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Lewis S, Clarke M (2001) Forest plots: trying to see the wood and the trees. BMJ 322:1479–1480
Xu XS, Yuan M, Zhu H, Yang Y, Wang H, Zhou H, Xu J, Zhang L, Pinheiro J (2018) Full covariate modelling approach in population pharmacokinetics: understanding the underlying hypothesis tests and implications of multiplicity. Br J Clin Pharmacol 84:1525–1534
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28:507–532
Gibiansky L, Gibiansky E, Kakkar T, Ma PP (2008) Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn 35:573–591
Long A, Chigutsa E, Wallin J (2017) Population pharmacokinetics of necitumumab in cancer patients. Clin Pharmacokinet 56:505–514
Ma P, Yang BB, Wang YM, Peterson M, Peterson M, Narayanan A, Sutjandra L, Rodriguez R, Chow AA (2009) Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J Clin Pharmacol 49:1142–1156
Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J (2018) Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: a comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MABS 10:751–764
Lobo ED, Hansen RJ, Balthasar JP (2004) Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 93:2645–2668
Yan X, Chan Y, Krzyzanski W (2012) Methods of solving rapid binding target-mediated drug disposition model for two drugs competing for the same receptor. J Pharmacokinet Pharmacodyn 39:543–560
Gibiansky L, Gibiansky E (2010) Target-mediated drug disposition model for drugs that bind to more than one target. J Pharmacokinet Pharmacodyn 37:323–346
Koch G, Jusko WJ, Schropp J (2017) Target mediated drug disposition with drug–drug interaction, Part II: competitive and uncompetitive cases. J Pharmacokinet Pharmacodyn 44:27–42
Hutmacher M, Kowalski KG (2014) Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol 79:132–147
Ryman JT, Meibohm B (2017) Pharmacokinetics of monoclonal antibodies. CPT Pharmacomet Syst Pharmacol 6:576–588
Azzopardi N, Lecomte T, Ternant T, Boisdron-Celle M, Piller F, Morel A, Gouilleux-Gruart V, Vignault-Desvignes C, Watier H, Gamelin E, Paintaud G (2011) Cetuximab pharmacokinetics influences progression-free survival of metastatic colorectal cancer patients. Clin Cancer Res 17:6329–6337
Shah MA, Xu RH, Bang YJ, Hoff PM, Liu T, Herráez-Baranda LA, Xia F, Garg A, Shing M, Tabernero J (2017) HELOISE: phase IIIb randomized multicenter study comparing standard-of-care and higher-dose trastuzumab regimens combined with chemotherapy as first-line therapy in patients with human epidermal growth factor receptor 2-positive metastatic gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol 35:2558–2567
Glassman PM, Balthasar JP (2016) Physiologically based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J Pharmacokinet Pharmacodyn 43:427–446
Bruno R, Washington CB, Lu J-F, Lieberman G, Banken L, Klein P (2005) Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 56:361–369
Hu C, Zhou H (2008) An improved approach for confirmatory phase III population pharmacokinetic analysis. J Clin Pharmacol 48:812–822
Funding
This study was funded by Symphogen A/S, DK-2750, Ballerup, Denmark.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M. Kragh, L. Alifrangis, N.J. Skartved and R. Hald were full time employees of Symphogen A/S at the time of the analysis. R. Schoemaker and J.R. Wade received a consultancy fee for the work in the paper and for travel expenses during this work, Drs. S. Kopetz, C. Montague, and J. Tabernero are paid scientific consultants for Symphogen A/S as well as for other companies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alifrangis, L., Schoemaker, R., Skartved, N.J. et al. Population pharmacokinetics and covariate analysis of Sym004, an antibody mixture against the epidermal growth factor receptor, in subjects with metastatic colorectal cancer and other solid tumors. J Pharmacokinet Pharmacodyn 47, 5–18 (2020). https://doi.org/10.1007/s10928-019-09663-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-019-09663-2