
Abstract
The inhibitory effect of anti-obesity drugs on energy intake (EI) is counter-acted by feedback regulation of the appetite control circuit leading to drug tolerance. This complicates the design and interpretation of EI studies in rodents that are used for anti-obesity drug development. Here, we investigated a synthetic long-acting analogue of the appetite-suppressing peptide hormone amylin (LAMY) in lean and diet-induced obese (DIO) rats. EI and body weight (BW) were measured daily and LAMY concentrations in plasma were assessed using defined time points following subcutaneous administration of the LAMY at different dosing regimens. Overall, 6 pharmacodynamic (PD) studies including a total of 173 rats were considered in this evaluation. Treatment caused a dose-dependent reduction in EI and BW, although multiple dosing indicated the development of tolerance over time. This behavior could be adequately described by a population model including homeostatic feedback of EI and a turnover model describing the relationship between EI and BW. The model was evaluated by testing its ability to predict BW loss in a toxicology study and was utilized to improve the understanding of dosing regimens for obesity therapy. As such, the model proved to be a valuable tool for the design and interpretation of rodent studies used in anti-obesity drug development.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Drug therapy of obesity in man has been hampered by the development of drug tolerance [1]. In clinical studies, body weight (BW) typically drops by up to 10% within the first 6 months of drug treatment, but then reaches a plateau and returns to levels of the placebo control groups upon treatment cessation. For instance, this response pattern has been observed for rimonabant [2], sibutramine [3,4,5], a combination of fenfluramine + phentermine [6], and pramlintide [7,8,9].
Studies in rats with measurement of daily food intake (FI) and BW indicated that the limited effect of anti-obesity drugs on BW is related to a loss of FI inhibition [1]. Typically, FI is maximally reduced on the first few days after initiation of drug treatment, but then rapidly increases and within 2 weeks attains a plateau at levels similar to, or only slightly lower than those of vehicle-treated rats. Following withdrawal of the drug, a rebound phase can even be observed, in which animals consume more food than the vehicle control group, leading to a return of BW comparable to that of control animals. This phenomenon is common to a range of agents with different mechanisms of action, including drugs such as the serotonin-norepinephrine reuptake inhibitor sibutramine [10, 11], the serotonin-releasing agent fenfluramine [12], the melanocortin-4 receptor activator melanotan II [13], as well as peptide hormones such as leptin [14], amylin [15,16,17] and glucagon-like peptide 1 (GLP-1) [18] or the GLP-1 analogue liraglutide [11, 19]. Therefore, the underlying feedback mechanism may not be target-specific, but rather related to the tight regulation of FI aimed at maintenance of a stable BW [1]. Short-term feedback signals from the gastrointestinal tract (e.g., cholecystokinin, peptide tyrosine and GLP-1) promote satiety leading to meal termination, while long-term adiposity signals (leptin and insulin) regulate long-term energy homeostasis and BW [20]. The peptide hormone amylin is thought to transmit both satiety and adiposity signals [21].
In this study, we focus on data from an anti-obesity-drug development program for synthetic long-acting analogues of amylin (LAMY). Amylin is synthesized in pancreatic β-cells and is co-secreted with insulin in response to ingestion meal [21,22,23]. It activates specific receptors in the hindbrain area postrema to suppress glucagon release, gastric emptying and FI, leading to reductions in blood glucose and ultimately BW [23]. Upon chronic administration to normal or diet-induced obese (DIO) rats via subcutaneous minipump, amylin inhibited FI (albeit with the above-described phenomena of tolerance and rebound), and caused a specific reduction in fat mass while maintaining lean mass [15, 16, 24, 25]. In two of these studies [15, 16], amylin was also found to prevent the compensatory decrease in energy expenditure (EE) that is typically observed with BW loss. Instead, the EE (expressed as a function of total BW) was increased, which was attributed to a relative increase in lean body mass, which is metabolically more active. However, since the amylin-mediated effect on EE was much smaller than its effect on FI, the latter was regarded to be the primary cause of BW loss.
In our LAMY drug development program, amylin analogues were screened using short-term studies in lean rats and selected peptides were tested in longer-term studies in DIO rats. Energy intake (EI) and BW were used as physiological response and clinical response biomarkers, respectively [26]. The presence of feedback regulation of EI complicates the determination of drug properties such as the IC50 from the rat studies. In this case, mathematical modeling accounting for feedback mechanisms serves as a valuable tool for data interpretation that may improve compound selection, study design and human dose estimation. Several models for EI and its feedback regulation have been published, ranging from simple, descriptive turnover models [27] to complex, mechanistic models incorporating the effects of leptin in regulating EI and expenditure [28, 29]. A semi-mechanistic model of intermediate complexity has been reported recently, referred to here as the “Gennemark model” [30]. In this model, EI data from the vehicle control group serves as a set point or reference, and drug-induced deviation from the set point drives a feedback signal that counteracts the drug effect and restores EI to the set point. This model described EI data from food restriction studies in rats and humans as well as EI data from mice and rats dosed with appetite-suppressing drugs. While this demonstrates the broad applicability of the model, the data base for the model fitting (e.g. the number of dose groups) was limited and could not support the adequate estimation of all model parameters. Moreover, the model did not include BW, which is the main variable of interest in obesity drug development.
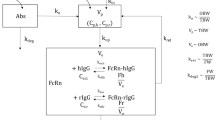
This article introduces a modified version of the Gennemark model that uses a more parsimonious homeostatic feedback function and additionally incorporates the relationship between EI and BW (Fig. 1). The model was developed based on rich data from lean and DIO rats treated with LAMY using various dosing regimens, which enabled an adequate estimation of all model parameters. We used a population approach to account for inter-individual variability and investigated whether model parameters differ between lean rats and DIO rats. Recognition of these differences would allow the prediction of drug effects in chronic DIO studies based on acute studies in lean rats that were used for compound-screening. We evaluated the model by testing its ability to predict a toxicology study that was not part of the model development dataset. Finally, we utilized the new model to understand how dosing regimens could be optimized for treatment of obesity.

Schematic representation of the PK/PD model. The PK/PD model consists of a PK model, an EI model and a BW model. The LAMY concentration–time profile is described by a 2-compartment model with first-order absorption from a depot compartment. The concentration in the central compartment inhibits EI via the drug inhibitory function I(C). This leads to a negative discrepancy relative to the reference EI without drug treatment, EIref. The resulting cumulative EI imbalance Y drives the appetite control signal h(Y7), which increases EI, thereby counter-regulating the drug inhibition. Several transit compartments (Y2–Y7) are included to describe the time delay between EI imbalance and homeostatic feedback. BW is characterized by a turnover model with EI as input and first-order BW loss. Variables for which data were available are indicated by grey shading. The model parameters are written in italics
Materials and methods
Rat studies
Ten independent preclinical studies using rats treated with LAMY are included in this publication. Studies 1, 8 and 9 were PK studies in Sprague–Dawley rats and Wistar rats, respectively. Studies 2–5 were single dose PD studies in lean Sprague–Dawley rats, studies 6 and 7 were longer-term, chronic PD studies in DIO Sprague–Dawley rats and study 10 was a 2-week toxicology study in lean Wistar rats (LAMY). An overview of the study details is given in Table 1. All animal experiments were conducted in accordance with internationally accepted animal welfare guidelines and were approved by the respective committees for animal research in Germany and Denmark. Male Sprague–Dawley rats were obtained from Taconic A/S, Denmark (studies 1–4 and 6) or Janvier Labs (studies 5 and 7–10). The animals were allowed to acclimatize for at least 7 days to their new environment before entering a study. The rats were housed in groups of n = 2 at 20–22 °C, relative humidity of 45–65% (study 1), 50–80% (studies 2–4 and 6) or 55 ± 10% (studies 5 and 7–10) and a 12:12 h light–dark cycle. During each study, animals had access to food and water ad libitum. In studies 1–5 and 8–10 normal chow was used (studies 1–4: Altromin 1324, Brogaarden A/S, Gentofte, Denmark; study 5 and 8–10: Diet 3430, Kliba Nafag, Provimi Kliba AG, Kaiseraugst, Switzerland). In studies 6 and 7, rats received a high-fat diet (HFD, study 6: 60% of total energy from fat, 58126 DIO Rodent Purified, Herfølge, Denmark; study 7: 60% of total energy from fat, D12492 Ssniff Spezialdiäten GmbH, Soest, Germany) for 12 or 20 weeks, respectively, prior to dosing and throughout the treatment period. The LAMY was freshly formulated prior to dosing using 50 mM histidine, 200 mM mannitol buffer pH 7 with and without 1% ammonia (studies 7 and 1–4, 6, respectively), 20 mM phosphate, 5% mannitol buffer pH 6 (studies 5, 8–10). In studies 6 and 7 animals received the vehicle on days with no LAMY administration. Drug concentration was analyzed in blood samples from defined time points. In PD studies 2–7, FI and BW were measured daily at the time of dosing. In all studies FI was assessed per cage, representing the FI of two rats (FI per rat was assumed to be half of the FI per cage).
Analysis of LAMY concentration in plasma
Blood samples of were collected in Microvette® tubes 0.50 mL K3EDTA (Sarstedt). The Microvette® tubes were ice-chilled prior to sampling. After collection, the blood was gently mixed by inverting the tube several times and stored upright on ice until centrifugation. Blood samples were centrifuged for 5 min at 8.300×g at 4 °C. After centrifugation, plasma was aliquoted and stored below − 70 °C until further analysis. Plasma concentrations of LAMY were analysed using a liquid chromatography/tandem mass spectrometry (LC/MS/MS) with an calibration range of 1–1000 nM for studies 1–4, 6 (Xevo TQ-S, Waters, Milford, Massachusetts, USA), and a range of 0.5–1000 for studies 5, 7–10 (QTrap 6500+, Sciex, Toronto, CA). Samples were pretreated with ethanol for protein precipitation before the analysis.
Modeling and simulation activities
Software, estimation methods and model evaluation
Model development and parameter estimation was performed using Phoenix WinNonlin 6.4, NLME 1.3 (Certara, Princeton, New Jersey, USA). Nonlinear mixed-effects (NLME) modeling was performed by applying the first-order conditional estimation–extended least squares (FOCE–ELS) with an interaction estimation method. A stepwise approach was used for model development. Firstly, the PK model was developed based on available plasma concentration data. Secondly, PD data (daily EI and BW) were modeled simultaneously using PK parameters estimated in step 1. This approach was chosen because there were many more PD observations compared to PK observations, and therefore the PK model could otherwise be affected by potential misspecifications in the PD model. Overall, BW was considered as the most important variable to assess the anti-obesity drug effect.
Simulations were performed by applying R, version 3.1.0 (R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org). In addition to the standard R packages, ggplot2, version 1.0.0 and deSolve, version 1.10-9 were used for plot generation and model-based simulations, respectively [31, 32].
Model selection and evaluation in all steps of model development were based on the following criteria: (a) successful model convergence (successful parameter estimation as well as estimation of the variance–covariance matrix), (b) numerical model selection criteria (− 2 times log likelihood (− 2LL), cut-off Δ3.84 for degrees of freedom, df = 1, representing α ≤ 0.05 for nested models), (c) precision of fixed and random parameter estimates (95% confidence interval of the respective parameter estimates did not include zero or one dependent on parameter inclusion), (d) standard goodness-of-fit plots (plots of observations versus (individual) predictions, individual predicted and observed time profiles, residual plots as well as ε- and η-distribution plots) and (e) visual-predictive checks, stratified by both study and dose group, but normalized for different starting BW. For visual predictive checks, simulations were performed 100 times for each subject and observation. The simulated time profiles were used to calculate the 95% confidence interval of the median time profiles, as well as the 80% prediction interval (10th–90th percentiles). For an adequate description of the data, the median of the observations should be within the calculated confidence interval and 80% of the observed data should be within the calculated 80% prediction interval of the simulated data.
The structural model
The PK/PD model describing the relationships between LAMY plasma concentration, EI and BW is illustrated in Fig. 1. The structural model consists of three parts, (a) the PK model for the LAMY compound, (b) the EI model, and (c) the BW turnover model.
The pharmacokinetic model
Different models ranging from 1- to 3-compartment models were investigated. In addition to the standard PK models, different absorption models with zero-order, parallel first-order absorption processes, and parallel first- and zero-order absorption processes were investigated. A two compartment model with first-order absorption was finally selected as the best PK model and used as a basis to evaluate the FI and BW model.
The food intake model
The second part of the structural model describes the FI and how the FI is inhibited by the LAMY compound. We assumed that the LAMY inhibits EI without influencing EE because the effect of amylin agonism on EE is not well investigated and seems to be negligible relative to the effect on EI [21,22,23]. Moreover, in-house indirect calorimetry studies did not show a significant change in EE following treatment with 2 nmol/kg LAMY every 2 days for 16 d, followed by an increased dose of 5 nmol/kg every 2 days for 14 d (data not shown). Therefore, EI was modeled as a function of LAMY plasma concentration by adapting an EI model with homeostatic feedback [30]. Energy intake was measured and modeled as cumulative EI (EI cum ) in kcal over a full day. In the model this was obtained by integration of Eq. (1) over 24 h (see Eqs. 1 and 2):
where EI(t) is energy intake at time t, EI ref represents the reference EI without drug treatment, I(C(t)) is the concentration-dependent inhibitory function at time t and h(Y 7 (t)) represents the appetite control signal at time t that will be defined below. Since our biomarker was EI cum over 24 h (i.e., daily measurements of EI), EI cum was calculated separately for each day by setting the initial condition to zero at the start of each day. Using continuous measurement of EI, we observed that rats feed primarily in the time from 5 pm to 5 am (dark phase 6 pm–6 am), consuming 2–3 major meals (data not shown). Therefore, EI ref was assumed to be constant for 12 h (starting 1 h prior to the dark phase) and was assigned to 0 for the remaining 12 h. This simple approach was used rather than a more accurate modeling of the complex pattern of EI ref because continuous measurements of EI were not performed in the reported studies. The circadian rhythm may be disregarded in case of constant drug concentrations (e.g., infusion), but should be considered when drug concentrations change significantly between the day and night phase. Since the LAMY compound is slowly absorbed, with peak plasma concentration (Cmax) reached after 24 h, we decided to incorporate the circadian rhythm of FI in order to accurately describe the FI, regardless if the drug is administered in the day or night phase.
I(C) was given by a sigmoid function of the plasma concentration C:
where I max is the maximum inhibitory potential, IC 50 is the LAMY concentration for 50% inhibition and n is the Hill exponent.
Feedback regulation of EI in the model is driven by the difference in EI with and without drug treatment. In order to consider not only the EI difference at time t, but also a memory of previous imbalances, a cumulative energy-imbalance function Y(t) was suggested by Gennemark et al. [30]. Here, Y(t) is obtained by integration of the ordinary differential equation:
The FI data indicated a delay before the cumulative energy imbalance elicits a feedback, potentially representing the time for turnover of signal mediators and/or signal transduction. Different numbers of transit compartments were empirically investigated to account for the delay in the feedback (range of transit compartments, n = 1–20). The best transit compartment model was selected as described in the model evaluation section. The equations for a transit feedback model with 7 transit compartments are described in Eqs. (5) and (6):
where k tr represents a first-order transit rate constant, and k represents the first-order rate constant describing the disappearance of the feedback signal from Y 7.
The appetite control signal h(Y 7) is a linear function of Y 7 with the empirical slope parameter h:
The body weight model
The third and last part of the structural model is the BW model. As the BW change over time directly depends on the EI over time, the parameters of this model were simultaneously estimated with the second part of the structural model (the FI model). The change in BW over time was modeled by a turnover model (Eq. 8), as previously described by [27]:
where EF represents the efficiency factor that describes conversion of energy (in kcal) to BW (in g) and k BW is a first-order rate constant describing the BW loss. The individual BW was initialized with the first measured BW for each rat. In contrast to the PK or EI model, the BW will not achieve steady-state because rats continue to grow throughout their life [33, 34]. As indicated earlier, the LAMY had no direct influence on energy conversion or relative BW loss; therefore, no drug effect terms were included in Eq. (8).
Equation (8) is a simplified version [35,36,37] of a mechanistic body composition model that has been used for mice [38,39,40], rats [41] and humans [35,36,37]. Our parameters EF and kBW correspond to 1/ρ and ε/ρ, respectively, where ρ and ε were previously defined by the following equations.
FM and FFM denote the 2 compartments fat mass and fat-free mass and α is a function describing the relationship between changes in FFM and FM. ρ is the energy density for changes in (F)FM, β is a parameter for diet-induced thermogenesis, γ is a proportionality constant for the relationship between metabolic rate and (F)FM, η is a proportionality constant for the relationship between energy expenditure and change in (F)FM and λ is a proportionality constant for physical activity per g (F)FM. Detailed descriptions of the body composition model [40] and its linearization [35, 37] are provided elsewhere.
The statistical model
The statistical model consists of two parts, the first one describing the inter-individual variability and potential inter-occasion variability, and the second part describing the residual (unexplained) variability. The inter-individual variability (IIV) was investigated in a stepwise procedure after each step of the model development (e.g., PK model, EI and BW model). Additional IIV terms were added subsequently according to the selection criteria described above (except the likelihood ratio test = drop in − 2LL). Inter-individual variability in rate constants was assumed to be log-normally distributed (to prevent negative parameter estimates, Eq. 11). For other model parameters, both log-normal and normal distributions of IIV were investigated (Eq. 12):
where θ k is the population estimate of the model parameter k, θ k,i is the individual parameter estimate for parameter k in individual i, and η k,i is the value of the random effect parameter on parameter k for the individual i. Inter-occasion variability could not be investigated as only one EI and BW data point was available for each individual per occasion, therefore no differentiation between inter-occasion variability and residual variability can be performed.
Different residual variability models (RV), accounting, e.g., for possible measurement errors, residual unexplained IIV, unexplained inter-occasional variability, and model misspecification, were investigated. Proportional (Eq. 13), additive (Eq. 14) and combined residual variability models (proportional + additive) were each investigated for plasma concentration, EI, and BW:
where y i,j is the jth observation for the ith individual, f i,j is the corresponding individual prediction and ε i,j is the residual variability term.
Covariate analysis
Potential covariate-parameter relations were pre-selected based on physiological plausibility, exploratory plots, as well as the data that were available for investigation. The covariates rat model (lean vs DIO), study type (PK vs PD studies) and investigator were tested on PK parameters. The covariates BW, rat model, study type, as well as food were additionally investigated on PD parameters. If none of these covariates could describe specific trends between and within studies, the study itself was investigated as a covariate to empirically quantify the difference to the other studies. The covariate-parameter relations were investigated as both proportional or power covariate-parameter relationships, using centering on the population mean in case of continuous covariates. Covariate effects were tested by forward selection and were included if they reduced the value of the -2LL by at least 3.84 points (α ≤ 0.05, assuming χ2-distribution of the difference in the − 2LL between the two nested models, 1 degree of freedom).
Results
LAMY pharmacokinetics
The PK of the LAMY in plasma was investigated in lean rats following i.v. and s.c. administration (study 1). As is typical for peptides [42], the i.v. PK was bi-exponential and was well-described by a 2-compartment PK model (Fig. 2, Table 2). The central volume of distribution (Vc) was 0.044 L/kg (95% confidence interval (CI): 0.0420–0.0460 L/kg), which is consistent with the plasma volume of Sprague–Dawley rats of comparable weight (0.041 L/kg [43]) and consistent with the distribution of the LAMY in plasma and extracellular fluid. The terminal half-life following s.c. administration in lean rats was 31 h, as desired for a long-acting peptide. The maximum concentration after s.c. administration was reached at 24 h post dose, due to a low absorption rate constant (ka) of 0.0334 h−1 (0.0315–0.0353 h−1).

LAMY Pharmacokinetics. Graphs show individual measured concentrations (symbols), as well as PK model fits (lines), following i.v. (grey) and s.c. (black) administration of 20 nmol/kg of the LAMY to lean rats (study 1)
LAMY concentrations were also measured in pharmacodynamic (PD) studies 2–7 following a range of doses, indicating dose-linearity of the PK (Fig. 3a). There was a trend for higher dose-normalized concentrations in DIO rats than in lean rats, which prompted us to do a covariate analysis on the disposition parameters V c , Cl, V p and Q. Plausible covariates would be the water content or fat-free mass since the distribution of peptides is usually confined to body water [42]. However, as these covariates were not measured and could not easily be estimated, we used the categorical covariate ‘rat model’. This covariate is also more convenient for the prediction of PK/PD studies without prior knowledge of the exact baseline body weights. The weight normalized volume and clearance parameters were significantly lower in DIO than in lean rats (52.6% (46.4–58.8%) and 22.6% (13.5–31.7%), respectively; Table 2). A comparable difference in PK parameters and resulting higher exposure in DIO rats versus lean rats was observed for another peptide drug, namely liraglutide (see Supplementary Fig. 1, Supplementary Table 1).

PK, EI and BW in an acute screening study in lean rats (study 2). Graphs and points show time profiles of plasma concentration (a), daily EI (b) and BW (c) in lean rats following a single s.c. dose of LAMY at 0 nmol/kg (light grey), 3 nmol/kg (medium grey), 10 nmol/kg (dark grey) and 30 nmol/kg (very dark grey). Symbols and whiskers represent a the mean ± SD for 2 animals (observed concentration), b and c the mean ± SD for 4 cages with 2 animals per cage (observed daily EI), or 8 animals (observed BW), respectively. d Hysteresis plot of concentration versus daily EI. Labels in d indicate time in days
There were also significant inter-study differences in the bioavailability (F) after s.c. administration. F was 0.489 (0.312–0.665), 0.582 (0.498–0.665) and 0.886 (0.798–0.972) in the PK study 1 and the PD studies 2–4 in lean rats at Zealand Pharma (ZP), the PD study 6 with DIO rats at ZP, and the PD studies 5 and 7 at Boehringer Ingelheim, respectively. Notably, a covariate effect on all systemic disposition parameters instead of F would describe the data equally well, but a change in F was thought to be more plausible (e.g., analytic bias, slightly different dose administered etc.). Overall, the model provided an adequate description of all PK data with exception of four outliers (Fig. 4a, Supplementary Figs. 2, 3).

Bland-Altman plots of residuals versus mean of individual predicted and observed values. Data are shown for plasma concentration (a), daily energy intake (EIcum) (b) and body weight (c) from i.v. PK studies (black), s.c. PK studies (grey), and PD studies (white). The dashed lines represent the mean ± the standard deviation, as well as the mean difference between the individual prediction and observation
Description of EI and BW data
The effect of the LAMY on EI and BW was tested in 5-day studies with a total of 109 lean rats (studies 2–5), and in chronic studies with a total of 64 DIO rats that had received a high-fat diet for several weeks prior to treatment (studies 6–7, see Table 1). In 5-day treatment studies, single LAMY doses, ranging from 3 to 200 nmol/kg, were administered s.c. to lean rats. This led to a dose-dependent decrease in daily EI and BW (shown exemplary for study 2 in Fig. 3b, c, also compare Fig. 5). EI was reduced most on day 2, but then returned to baseline faster than expected based on measured plasma LAMY concentrations (Fig. 3a, b), suggesting feedback regulation of EI. This becomes clear when comparing EI at two different time points with similar measured concentrations (e.g., 1 day after dosing of 10 nmol/kg and 4 days after dosing of 30 nmol/kg, Fig. 3a, b). The feedback regulation is also reflected in the concentration-EI relationships presented in Fig. 3d. EI inhibition resulted in BW loss relative to the vehicle group, which was maximal within 24 h from the maximum EI effect.

Visual predictive check for study 2 (lean rats treated with LAMY). Graphs show time profiles of daily EI (top) and BW (bottom) in lean rats receiving vehicle (a, e) or LAMY s.c. at 3 nmol/kg (b, f), 10 nmol/kg (c, g) or 30 nmol/kg (d, h). Symbols are individual data for 4 cages with 2 animals per cage (daily EI), or 8 animals (BW). BW data are normalized to initial BW. Black lines represent the median of observations while the dark grey shading indicates the simulation-based 95% confidence interval of the median. The 80% prediction interval (10th–90th percentiles) is shown as light grey shading
The anti-obesity effect of chronic LAMY treatment was investigated in DIO rats. A dosing interval of 2 days was used to measure effects with a low fluctuation of peptide concentration as expected for humans with weekly administration, while higher dosing intervals of 4 days were included to be able to observe the feedback-driven return of EI and BW to baseline. Overall treatment duration was ca. 4 weeks, a time frame that corresponds approximately to 1 year in humans when adjusting for average life span of the species [30]. The EI over time in DIO rats was comparable to that in lean rats, showing a quick initial drop in EI followed by a slow increase. After approximately 12 days, EI reached a steady state slightly lower than that of the vehicle group (shown for studies 6 and 7 in Fig. 6 and Supplementary Fig. 7, respectively). When comparing groups in studies 6 and 7 that received the same (10 nmol/kg every 4 days), one notices that the maximum EI reduction was achieved after 2 days in study 6 compared to a maximum FI inhibition 1 day after LAMY administration in study 7. Knowing the PK of LAMY and the circadian rhythm of FI, this can be explained by the different timing of LAMY administration in the two studies (see Table 1). In study 6, LAMY was administered directly at the start of the feeding phase (night phase). Since the drug is absorbed slowly (tmax = 24 h), maximum plasma concentrations and hence inhibition of FI was not achieved within the first feeding phase. In study 7, LAMY concentrations during the feeding phase on the first day were higher since the LAMY compound was administered 8 h before the feeding phase. This also demonstrates the importance of applying a dose–exposure–response analysis, as a standard dose–response analysis would not be sufficient to describe this time-dependent food inhibition.

Visual predictive check for study 6 (DIO rats treated with LAMY). Graphs show time profiles of daily EI (top) and BW (bottom) in DIO rats receiving vehicle (a, e) or LAMY s.c. at 1 nmol/kg every 2 days (b, f), 3 nmol/kg every 2 days (c, g) or 10 nmol/kg every 4 days (d, h). Symbols are individual data for 4 cages with 2 animals per cage (EI plots) and 8 animals (BW plots). BW data are normalized to initial BW. Color coding is as in Fig. 5
BW initially decreased relative to the vehicle group and reached the lowest values ca. 11 days following maximum EI inhibition, but did not drop further in the last 2 weeks of treatment (Fig. 6). The vehicle-corrected BW loss was 5.77% (1 nmol/kg every 2 days), 10.1% (3 nmol/kg every 2 days) and 16.3% (10 nmol/kg every 4 days) in study 6, and to 11% (3 nmol/kg every 2 days) and 10% (10 nmol/kg every 4 days) in study 7 (Supplementary Fig. 7). Overall, the results indicate the presence of a feedback regulation of EI that gives rise to tolerance against the LAMY with multiple dosing.
Modeling of EI and BW
We investigated whether the EI and BW data can be described by a PK/PD model that integrates drug-mediated inhibition of EI and counter-modulatory feedback (Eqs. 1–8). The parameters of the model were estimated simultaneously based on data from studies 2–7. The incorporation of the circadian rhythm of FI was necessary for the model to describe the observed dependence of the time to maximum EI reduction on the time period between drug administration and the feeding phase. A good description of the initial EI reduction enabled an adequate estimation of the model parameters, especially the IC50.
The feedback signal in our model is a linear function of the difference in cumulative EI between treated and untreated rats (Eqs. 6, 7). It includes only a single parameter h and is therefore more parsimonious than the previously used Gompertz function with the upper asymptote h max and the slope parameter h slope [30]. While the Gompertz function was investigated, it did not provide any statistical superiority. The feedback function was added to, rather than multiplied with [30] EIref (Eq. 1) because the latter could not properly describe an initial EI inhibition > 90% followed by feedback-mediated EI increase. A time delay of the feedback signal in the form of transit compartments was required to adequately describe the strong initial reduction in EI in combination with a feedback after the first days. The parameter estimate for k tr was 0.277 h−1 (0.248–0.305 h−1), corresponding to a mean transit time of 25.3 h for onset of the feedback.
IIV terms were tested for all PD parameters, but the data could only support IIV estimation for EI without drug treatment during the feeding phase (EI ref,on ) and the IC 50. A plot of η EI,ref,on separated by study revealed inter-study-differences in EI ref,on , which could partly be described by a covariate effect of 7.27% (3.40–11.1%) higher EI in DIO rats than in lean rats. This is in agreement with previous studies comparing EI in male SD rats fed standard and high-fat diets [44, 45]. Furthermore, the data indicated that EI ref,on in study 5 differed from the other studies in lean rats with 24.8% (15.7–33.8%) higher EI. Potential reasons could be the different diet source, animal provider or study site used in study 5 (see Table 1).
When adding an IIV parameter on the first-order rate constant describing the BW loss (k BW ) instead of the IC 50 , plots of observations versus population predictions indicated a model misspecification especially for rats with very high BW. This trend could partly be described by a power covariate-parameter relationship (see footnote c of Table 3), which assigns lower values of k BW,i to individuals with higher BW. The population estimate of k BW (0.00169 h−1 (0.00140–0.00199 h−1)) is equivalent to a half-life of 17.1 days (representing the time to 50% weight loss in the absence of FI). IIV on k BW was not retained in the final model because prediction versus observation plots indicated a better data description when adding IIV on IC 50 instead of k BW .
All fixed and random effect parameters of the model were estimated with acceptable precision with relative standard errors (RSE) ≤ 20% (see Tables 2, 3), except the covariate influences of the rat type on EI (RSE = 27.2%) and the higher F in study 6 (RSE = 45.4%). The model performance was evaluated using Bland–Altman plots (Fig. 4) and standard goodness-of-fit plots (Supplementary Figs. 2, 3). In addition, visual predictive checks were performed (Figs. 5, 6, Supplementary material Figs. 4, 5, 6, 7).
Overall the model provided an adequate description of the data, although the median of the EI observations lay below the calculated 95% confidence interval of the simulation-based median in Fig. 6c and d. This indicates that the speed of the feedback on EI within individual dose groups was overestimated. Moreover, the model could not describe the trend for a study-specific time-dependent decrease (Fig. 6) or increase (Supplementary Fig. 7) of EI in the vehicle control group. As no reason for this inter-study discrepancy was identified, this phenomenon was not included in the model. Therefore, the highest EI values (representing reference EI at the end of study 7) were under-predicted (see Fig. 4b). However, the model residuals of the PK and BW model appeared randomly and approximately normally distributed (Supplementary Fig. 3), as expected for an adequately specified mathematical model [46]. The most important readout, the BW, was well described over the whole range of predicted BW (see Fig. 4c).

Comparison of LAMY PK/PD between lean rats and DIO rats. a, b Simulated time profiles of concentration (a) and reference-normalized daily EI (b) and BW (c) for DIO (black lines) and lean rats (grey lines) dosed with 10 nmol/kg every 2 days for 4 weeks, followed by 2 weeks without treatment. Dotted lines indicate levels in the vehicle control group. Simulations used the PK/PD population estimates given in Tables 2, 3. d Steady-state relationship between the average concentration (Cave) and the average EI (EIave) within one dose interval. The steady-state relationship is identical for lean and DIO rats
The impact of the differences in PK parameters and k BW between lean and DIO rats was illustrated by simulating a 4-week treatment with 10 nmol/kg LAMY every 2 days, followed by a 2-week washout phase (Fig. 7a–c). The simulation shows higher plasma concentrations in DIO than lean rats, leading to a slightly higher initial EI reduction and thus faster BW loss. The model predicts a rebound of EI following treatment discontinuation that is consistent with observations in the literature (see for example [1]). The steady-state relationship between average LAMY concentration (Cave) and EI (EIave) during treatment was determined by simulating 12 doses in a 1000-fold dose range (Cave and EIave were used for the steady-state relationship to be applicable to different dose intervals). This showed that the Cave required at steady state for a 10% reduction of EIave is ~ 30 nmol/L (Fig. 7d). At average concentrations > 100 nmol/L EIave approached a lower asymptote with approximately 15% reduction relative to the vehicle-treated reference group.
Model-based design of a toxicology study
Dose selection for toxicology studies is a balancing act for obesity drugs because doses need to be sufficiently high to examine adverse effects, but should not elicit BW loss > 20% relative to the vehicle group according to animal ethics regulations. Therefore, we used the LAMY PK/PD model to plan a toxicology study with lean Wistar rats. The PK parameters were determined from i.v. and s.c. LAMY PK studies in Wistar rats to account for strain differences in PK between Wistar and Sprague–Dawley rats (see Supplementary Table 2). The PD parameters were assumed to be equal in the two rat strains although there may well be differences in growth rate or feedback. Dosing three times a week with 2, 6, 20 and 60 nmol/kg was predicted to cause BW loss of 3.28, 9.52, 16.2 and 19.1%, respectively, relative to the vehicle group. The data were in broad agreement with these predictions (Fig. 8). The model correctly predicted the saturation of BW loss at the two highest dose levels, although the maximum BW loss in these two groups was slightly over-predicted (possibly due to an under-estimation of homeostatic feedback).

Prediction of BW in a toxicology study (study 10). The graph shows predictions (lines) and mean BW (symbols) of Wistar rats dosed with LAMY s.c. thrice weekly (Monday, Wednesday and Friday), as indicated by the vertical black lines, with 0 nmol/kg (black), 2 nmol/kg (dark grey), 6 nmol/kg (medium grey), 20 nmol/kg (grey) and 60 nmol/kg (light grey). All data are normalized to the mean BW of the vehicle group
Simulation of dose escalation
Using model-based simulations, we explored the utility of escalating dosing schemes, because these are often used to increase tolerability in rat toxicology studies and in the clinic (e.g., for liraglutide [47] and pramlintide [48]). As shown in Fig. 9, a dose of 9 nmol/kg causes a higher reduction of EI in treatment-naïve rats than in rats previously dosed for 12 days with lower doses of 3 nmol/kg and 6 nmol/kg every 2 days. In the latter case a higher cumulative energy imbalance and thus a higher feedback signal is already in place at the time of the first 9 nmol/kg dose (on day 12) due to previous energy imbalances (Fig. 9c). This simulation exercise suggests that dose escalation may be a reasonable strategy for clinical trials. It could be applied if the FI reduction was too strong in a single rising dose Phase I study but higher doses were required to maintain sufficient efficacy also for chronic treatment.

PK/PD simulation illustrating the effect of dose escalation. Time profiles of concentration (a), reference-corrected daily EI (b), the cumulative energy imbalance Y7 (c) and reference-corrected BW (d) from a simulation using the PK/PD parameters for DIO rats from Tables 2 and 3. Grey line: Dosing with 9 nmol/kg every 2 days. Black line: Dose escalation, dosing every 2 days with 3 nmol/kg (doses 1–3), 6 nmol/kg (doses 4–6) and 9 nmol/kg (doses 7–14). Vertical dotted lines indicate the different dosing periods
Discussion
Data on the effect of a LAMY on EI and BW in lean and DIO rats are presented. Similar to amylin [15, 16, 24] and other appetite-suppressing drugs [1], the LAMY used showed a pronounced initial reduction of EI that levelled off to a steady-state with EI slightly lower than in the vehicle group upon chronic treatment (Fig. 6, Supplementary Fig. 7). This biphasic response pattern was overall well-described by a modified version of a previous PK/PD model accounting for homeostatic feedback of EI [30].
In contrast to the previous publication [30], we used a larger data base for a single compound (different doses and dosing intervals), which allowed the estimation of all model parameters with adequate precision (Tables 2, 3). The ability of the model to integrate lean and DIO rat data from seven different studies using different dosing schedules increased the confidence in the predictive power of the model. Inter-individual variability was accounted for by using a population approach. This is especially useful for modeling of highly variable BW data, because the shape of the typical BW time profile may be very different from individual BW time profiles. The model predicted a steady-state relationship between Cave and EIave with a lower asymptote of 15% reference-corrected reduction, which is in contrast to the greater EI reduction (35%) predicted by the previous model at high concentrations [30] (a lower asymptote was not shown in the respective publication). This is due to the use of a linear function in place of the saturable Gompertz function previously used to account for physiological limits of feedback regulation [30]. There will be an upper physiological limit for the feedback, but its determination would require prolonged fasting (e.g., by using high LAMY doses), causing greater weight loss than allowed by animal ethics considerations. In the absence such data, the presented linear function provides a good mathematical approximation of the observed effects.
Model predictions deviated from observations mainly in two respects. Firstly, the model did not describe the study-specific time-dependent change of EI in vehicle-treated DIO rats (Fig. 6a, Supplementary Fig. 7). This could be considered in the model by assuming that the baseline EI, EI ref , is not constant, but a linear function of time. However, the opposing trends in EI observed in studies 6 and 7 would have required the additional inclusion of covariate and/or random effects, which was not supported by the available data. Secondly, the observed increase in EI following maximum EI reduction in DIO rats was slower than predicted (see Fig. 6). A reason might be that rats ate less due to nausea, which is a common adverse event of amylin- and GLP1-analogues in the clinic [8, 47], and has also been observed in rats treated with these agents [49, 50]. Nausea was not accounted for in the model, because its underlying mechanism as well as its dose and time dependence are poorly understood. An alternative reason may be a combination of feedback mechanisms with different time scales that are not described by the model with only a single feedback mechanism included. In the future, it would be useful to discriminate short-term satiation feedback and long-term adiposity feedback [20]. This however, would require time-resolved data of the relevant hormone levels in plasma.
In addition to the discrepancies discussed above, the model has the following limitations. Firstly, a complete understanding of the PK is limited by the sparsity of plasma LAMY concentration data—especially for DIO rats. Therefore, reasons for the observed inter-study variability in PK cannot be identified easily. Secondly, the model does not account mechanistically for potential drug-mediated changes in homeostatic feedback. For instance, there is evidence that amylin agonism restores sensitivity to leptin [9], which is a peptide hormone secreted by adipose cells in proportion to adipose tissue mass and regulates BW by inhibiting FI and increasing energy expenditure [51]. Thus, feedback stimulation of FI may decrease with time of LAMY treatment, although there was no indication of this in our data. Finally, our descriptive BW model simplifies the relationship between EI and BW. More mechanistic models for body composition of mice [38,39,40], rats [41] and humans [35,36,37] have been published previously. These models consider the two compartments fat mass (FM) and fat-free mass (FFM) with different energy densities, whose weight changes are driven by the difference between EI and EE. EE in the models accounts for physical activity and metabolic rate (and their dependence on FM and FFM) as well as basal and diet-induced thermogenesis and the energy required for degradation of fat or protein. A key assumption of the body composition model is that the relationship between changes in FFM and FM can be described by a time-invariant Forbes function α. The model by Selimkhanov et al. [41], that was developed for rats of the same strain and gender as used in our studies, assumed that α is constant. In this case the body composition model can be simplified by introducing the parameters ρ and ε ([35,36,37], see Eqs. 9 and 10). This yields the one-compartment BW model used by us (see Eq. 8) where EF and kBW equal 1/ρ and ε/ρ, respectively. The values of EF (0.101 g/kcal) and kBW (0.000937 h−1) as calculated from the parameters of the rat body composition model [41] are similar to our fitted estimates of EF (0.272 g/kcal) and kBW (0.00169 h−1). However, the values are not directly comparable because we included an additional parameter to consider the observed decrease of individual kBW with increasing BW (COVBW,kBW). This covariate effect was required for an adequate description of the data and may have accounted for effects not incorporated in the mechanistic model (e.g., lower physical activity or better insulation of the body in obesity). Moreover, it may have compensated for the crude and restrictive assumption of a constant α value. Selimkhanov et al. already noticed that their model with a constant α did not adequately describe body composition of rats treated with a drug that impacted FM more than FFM. LAMY would also fall into that category, since amylin agonism is known to specifically reduce FM while maintaining FFM [15, 16, 24, 25]. Therefore, the use of the Selimkhanov model would not have improved the description of our BW data. However, future rat studies that include the measurement of EE and fat mass may allow the development of more suitable mechanistic body composition models.
Despite its limitations, our model provided an adequate description of the BW of rats ranging from 250 to 700 g, over time scales typically used in preclinical proof-of-concept studies. The model may therefore be valuable for model-informed study design.
Our PK/PD model was developed based on data for both lean and DIO rats. The simultaneous modeling of lean and DIO rat data allowed identifying differences in parameters between the two rat models. The weight-normalized volume and clearance parameters of the tested LAMY were lower in DIO than in lean rats (Table 2). The validity of this result is limited by the sparse exposure data for DIO rats and the lack of an intra-study comparison of LAMY PK in lean and DIO rats, but was corroborated by liraglutide PK data (Supplementary Table 1, Supplementary Fig. 1). Lower weight-normalized disposition parameters for DIO rats are in line with theoretical considerations, since peptides generally distribute in body water [42], but lean and DIO rats differ more in fat mass than in water content. Since these considerations are also applicable to humans, the BW dependence of the PK should be investigated in clinical studies with our LAMY. BW was previously found to be a predictor of the apparent clearance (Cl/F) of liraglutide in patients with type 2 diabetes or obesity [52,53,54]. In these studies the covariate effect on Cl/F was described by an allometric relationship with exponent < 1, which is in line with a decrease of weight-normalized Cl with BW. Regarding the parameters of the BW model, the modeling identified a dependence of individual kBW on BW (discussed above), but no difference between EF in lean and DIO rats. For both rat types, EF was estimated to be 0.272 g/kcal. This means that approximately 3.7 kcal are required to gain 1 g of BW. This value is between the estimated values of energy density for changes in FM (9.4 kcal/g) and FFM (1.8 kcal/g) in rat [55].
The knowledge of the parameter differences and similarities between lean and DIO rats can help in the design of DIO studies based on acute studies in lean rats that are earlier in the screening cascade of drug discovery. The data from chronic DIO studies may then be used to refine the model and to plan toxicology studies that are typically performed in lean rats. The utility of this approach is illustrated by the adequate prediction of a toxicology study using the LAMY PK/PD model (Fig. 8). This approach does not necessarily require the amount of data reported in this manuscript. Based on our experience and exploratory runs, the following studies appear sufficient to inform an adequate model: (1) a single dose PD study in lean rats, using 3 different doses that span the range from low to high EI effects; (2) a chronic PD study with 4-week multiple dosing in DIO rats, using three different doses (and different dose intervals if possible); and (3) a single dose PK study in both lean and DIO rats. Ideally, both PD studies should include the washout phase, since data on the rebound of EI and the resulting BW gain contain valuable information about the feedback and the BW model. Our simulation showed that a study extension by ca. 10 days would suffice to observe the rebound phase (Fig. 7). This would facilitate model parameterization with little additional expense, especially when using automatic recording of FI and limiting BW measurements to two or three times weekly. Once the model is calibrated (combined with an external evaluation for a second compound), it may be sufficient to perform a single dose PD study in lean rats per compound (again ideally including the washout phase). By modeling these data, different drug candidates can be ranked according to their in vivo IC50, which cannot be estimated otherwise (see Fig. 3d). Using the determined IC50 values, the efficacy in DIO rats may then be predicted to enable selection of only the most promising candidates, at the most suitable dosing regimens, for subsequent testing in rats.
Our model was also used for simulations to identify dosing schemes that may be useful for preclinical or clinical studies—provided that the model translates to human at least qualitatively. This showed that a step-wise dose escalation can be used to avoid a high initial EI reduction even though this delays BW loss (Fig. 9). The more uniform EI inhibition achieved with dose escalation may be useful to avoid adverse effects possibly associated with strong EI inhibition in rats and humans, and to increase patient compliance in the clinic.
The ultimate goal would be the translation of our model to man in order to predict the efficacious dose in the clinic and to determine optimal dosing regimens (see for example [56]). The PK of peptides can usually be translated to man using allometric scaling or using existing clinical PK data from comparable peptides. The BW model for the rat could be replaced by the corresponding human model [35,36,37]. The question how feedback mechanisms translate to human would be the most challenging one. In order to answer this question, modeling of data from rodent and clinical studies with the same compound would be required. To our knowledge such a comparison has not been performed so far. In addition to homeostatic feedback considered in our model, hedonic and cognitive feedback circuits may also play a role in man [57]. In the absence of more detailed knowledge on feedback mechanisms, the effective concentrations determined in rodents can provide a reasonable target concentration in human (comparable affinity of LAMY to the human and rat receptor, data not shown here).
Conclusions
We showed that a LAMY can reduce EI and BW in rats, although homeostatic feedback of EI leads to tolerance with multiple dosing. This behavior was adequately described by a population PK/PD model including homeostatic feedback for EI, and an EI-dependent turnover model to describe the BW change over time. The model was evaluated by predicting the expected BW in a separate toxicology study. Furthermore, the model provides the opportunity to extrapolate from lean single dose studies to multiple dose DIO rat studies in drug development. In summary, the model may be valuable for the design and interpretation of rodent studies with anti-obesity drugs.
References
Fernstrom JD, Choi S (2008) The development of tolerance to drugs that suppress food intake. Pharmacol Ther 117(1):105–122. https://doi.org/10.1016/j.pharmthera.2007.09.001
Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J (2006) Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: a randomized controlled trial. JAMA 295(7):761–775. https://doi.org/10.1001/jama.295.7.761
Bray GA, Ryan DH, Gordon D, Heidingsfelder S, Cerise F, Wilson K (1996) A double-blind randomized placebo-controlled trial of sibutramine. Obes Res 4(3):263–270
James WP, Astrup A, Finer N, Hilsted J, Kopelman P, Rossner S, Saris WH, Van Gaal LF (2000) Effect of sibutramine on weight maintenance after weight loss: a randomised trial. STORM Study Group. Sibutramine trial of obesity reduction and maintenance. Lancet 356(9248):2119–2125
Wirth A, Krause J (2001) Long-term weight loss with sibutramine: a randomized controlled trial. JAMA 286(11):1331–1339
Weintraub M (1992) Long-term weight control: The National Heart, Lung, and Blood Institute funded multimodal intervention study. Clin Pharmacol Ther 51(5):581–585
Ravussin E, Smith SR, Mitchell JA, Shringarpure R, Shan K, Maier H, Koda JE, Weyer C (2009) Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity (Silver Spring) 17(9):1736–1743. https://doi.org/10.1038/oby.2009.184
Smith SR, Aronne LJ, Burns CM, Kesty NC, Halseth AE, Weyer C (2008) Sustained weight loss following 12-month pramlintide treatment as an adjunct to lifestyle intervention in obesity. Diabetes Care 31(9):1816–1823. https://doi.org/10.2337/dc08-0029
Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, Baron AD (2008) Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci USA 105(20):7257–7262. https://doi.org/10.1073/pnas.0706473105
Fisas A, Codony X, Romero G, Dordal A, Giraldo J, Merce R, Holenz J, Vrang N, Sorensen RV, Heal D, Buschmann H, Pauwels PJ (2006) Chronic 5-HT6 receptor modulation by E-6837 induces hypophagia and sustained weight loss in diet-induced obese rats. Br J Pharmacol 148(7):973–983. https://doi.org/10.1038/sj.bjp.0706807
Madsen AN, Hansen G, Paulsen SJ, Lykkegaard K, Tang-Christensen M, Hansen HS, Levin BE, Larsen PJ, Knudsen LB, Fosgerau K, Vrang N (2010) Long-term characterization of the diet-induced obese and diet-resistant rat model: a polygenetic rat model mimicking the human obesity syndrome. J Endocrinol 206(3):287–296. https://doi.org/10.1677/JOE-10-0004
Choi S, Jonak EM, Simpson L, Patil V, Fernstrom JD (2002) Intermittent, chronic fenfluramine administration to rats repeatedly suppresses food intake despite substantial brain serotonin reductions. Brain Res 928(1–2):30–39
Seeley RJ, Burklow ML, Wilmer KA, Matthews CC, Reizes O, McOsker CC, Trokhan DP, Gross MC, Sheldon RJ (2005) The effect of the melanocortin agonist, MT-II, on the defended level of body adiposity. Endocrinology 146(9):3732–3738. https://doi.org/10.1210/en.2004-1663
Sahu A (2002) Resistance to the satiety action of leptin following chronic central leptin infusion is associated with the development of leptin resistance in neuropeptide Y neurones. J Neuroendocrinol 14(10):796–804
Roth JD, Hughes H, Kendall E, Baron AD, Anderson CM (2006) Antiobesity effects of the beta-cell hormone amylin in diet-induced obese rats: effects on food intake, body weight, composition, energy expenditure, and gene expression. Endocrinology 147(12):5855–5864. https://doi.org/10.1210/en.2006-0393
Mack C, Wilson J, Athanacio J, Reynolds J, Laugero K, Guss S, Vu C, Roth J, Parkes D (2007) Pharmacological actions of the peptide hormone amylin in the long-term regulation of food intake, food preference, and body weight. Am J Physiol Regul Integr Comp Physiol 293(5):R1855–R1863. https://doi.org/10.1152/ajpregu.00297.2007
Roth JD, Hughes H, Coffey T, Maier H, Trevaskis JL, Anderson CM (2007) Effects of prior or concurrent food restriction on amylin-induced changes in body weight and body composition in high-fat-fed female rats. Am J Physiol Endocrinol Metab 293(4):E1112–E1117. https://doi.org/10.1152/ajpendo.00395.2007
Donahey JC, van Dijk G, Woods SC, Seeley RJ (1998) Intraventricular GLP-1 reduces short- but not long-term food intake or body weight in lean and obese rats. Brain Res 779(1–2):75–83
Larsen PJ, Fledelius C, Knudsen LB, Tang-Christensen M (2001) Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes 50(11):2530–2539
Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW (2006) Central nervous system control of food intake and body weight. Nature 443(7109):289–295. https://doi.org/10.1038/nature05026
Lutz TA (2012) Effects of amylin on eating and adiposity. Handb Exp Pharmacol 209:231–250. https://doi.org/10.1007/978-3-642-24716-3_10
Lutz TA (2012) Control of energy homeostasis by amylin. Cell Mol Life Sci 69(12):1947–1965. https://doi.org/10.1007/s00018-011-0905-1
Hay DL, Chen S, Lutz TA, Parkes DG, Roth JD (2015) Amylin: pharmacology, physiology, and clinical potential. Pharmacol Rev 67(3):564–600. https://doi.org/10.1124/pr.115.010629
Roth JD, Coffey T, Jodka CM, Maier H, Athanacio JR, Mack CM, Weyer C, Parkes DG (2007) Combination therapy with amylin and peptide YY[3-36] in obese rodents: anorexigenic synergy and weight loss additivity. Endocrinology 148(12):6054–6061. https://doi.org/10.1210/en.2007-0898
Isaksson B, Wang F, Permert J, Olsson M, Fruin B, Herrington MK, Enochsson L, Erlanson-Albertsson C, Arnelo U (2005) Chronically administered islet amyloid polypeptide in rats serves as an adiposity inhibitor and regulates energy homeostasis. Pancreatology 5(1):29–36. https://doi.org/10.1159/000084488
Danhof M, Alvan G, Dahl SG, Kuhlmann J, Paintaud G (2005) Mechanism-based pharmacokinetic-pharmacodynamic modeling-a new classification of biomarkers. Pharm Res 22(9):1432–1437. https://doi.org/10.1007/s11095-005-5882-3
Fang J, DuBois DC, He Y, Almon RR, Jusko WJ (2011) Dynamic modeling of methylprednisolone effects on body weight and glucose regulation in rats. J Pharmacokinet Pharmacodyn 38(3):293–316. https://doi.org/10.1007/s10928-011-9194-4
Jacquier M, Crauste F, Soulage CO, Soula HA (2014) A predictive model of the dynamics of body weight and food intake in rats submitted to caloric restrictions. PLoS One 9(6):e100073. https://doi.org/10.1371/journal.pone.0100073
Tam J, Fukumura D, Jain RK (2009) A mathematical model of murine metabolic regulation by leptin: energy balance and defense of a stable body weight. Cell Metab 9(1):52–63. https://doi.org/10.1016/j.cmet.2008.11.005
Gennemark P, Hjorth S, Gabrielsson J (2015) Modeling energy intake by adding homeostatic feedback and drug intervention. J Pharmacokinet Pharmacodyn 42(1):79–96. https://doi.org/10.1007/s10928-014-9399-4
Ito K, Murphy D (2013) Application of ggplot2 to pharmacometric graphics. CPT 2:e79. https://doi.org/10.1038/psp.2013.56
Soetaert K, Petzoldt T, Setzer RW (2010) Solving differential equations in R: package deSolve. J Stat Softw 33(9):1–25. https://doi.org/10.1137/0915088
Newby FD, DiGirolamo M, Cotsonis GA, Kutner MH (1990) Model of spontaneous obesity in aging male Wistar rats. Am J Physiol 259(6 Pt 2):R1117–R1125
Hubert MF, Laroque P, Gillet JP, Keenan KP (2000) The effects of diet, ad Libitum feeding, and moderate and severe dietary restriction on body weight, survival, clinical pathology parameters, and cause of death in control Sprague-Dawley rats. Toxicol Sci 58(1):195–207
Chow CC, Hall KD (2008) The dynamics of human body weight change. PLoS Comput Biol 4(3):e1000045. https://doi.org/10.1371/journal.pcbi.1000045
Polidori D, Sanghvi A, Seeley RJ, Hall KD (2016) How strongly does appetite counter weight loss? Quantification of the feedback control of human energy intake. Obesity (Silver Spring) 24(11):2289–2295. https://doi.org/10.1002/oby.21653
Chow CC, Hall KD (2014) Short and long-term energy intake patterns and their implications for human body weight regulation. Physiol Behav 134:60–65. https://doi.org/10.1016/j.physbeh.2014.02.044
Guo J, Hall KD (2011) Predicting changes of body weight, body fat, energy expenditure and metabolic fuel selection in C57BL/6 mice. PLoS ONE 6(1):e15961. https://doi.org/10.1371/journal.pone.0015961
Guo J, Hall KD (2009) Estimating the continuous-time dynamics of energy and fat metabolism in mice. PLoS Comput Biol 5(9):e1000511. https://doi.org/10.1371/journal.pcbi.1000511
Gennemark P, Jansson-Lofmark R, Hyberg G, Wigstrand M, Kakol-Palm D, Hakansson P, Hovdal D, Brodin P, Fritsch-Fredin M, Antonsson M, Ploj K, Gabrielsson J (2013) A modeling approach for compounds affecting body composition. J Pharmacokinet Pharmacodyn 40(6):651–667. https://doi.org/10.1007/s10928-013-9337-x
Selimkhanov J, Thompson WC, Patterson TA, Hadcock JR, Scott DO, Maurer TS, Musante CJ (2016) Evaluation of a mathematical model of rat body weight regulation in application to caloric restriction and drug treatment studies. PLoS ONE 11(5):e0155674. https://doi.org/10.1371/journal.pone.0155674
Diao L, Meibohm B (2013) Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet 52(10):855–868. https://doi.org/10.1007/s40262-013-0079-0
Probst RJ, Lim JM, Bird DN, Pole GL, Sato AK, Claybaugh JR (2006) Gender differences in the blood volume of conscious Sprague-Dawley rats. J Am Assoc Lab Anim Sci 45(2):49–52
Marques C, Meireles M, Norberto S, Leite J, Freitas J, Pestana D, Faria A, Calhau C (2016) High-fat diet-induced obesity Rat model: a comparison between Wistar and Sprague-Dawley Rat. Adipocyte 5(1):11–21. https://doi.org/10.1080/21623945.2015.1061723
Gong H, Han YW, Sun L, Zhang Y, Zhang EY, Li Y, Zhang TM (2016) The effects of energy intake of four different feeding patterns in rats. Exp Biol Med (Maywood) 241(1):52–59. https://doi.org/10.1177/1535370215584890
Owen JS, Fiedler-Kelly J (2014) Introduction to population pharmacokinetic/pharmacodynamic analysis with nonlinear mixed effects models. Wiley, Hoboken
Lean ME, Carraro R, Finer N, Hartvig H, Lindegaard ML, Rossner S, Van Gaal L, Astrup A, Investigators NN (2014) Tolerability of nausea and vomiting and associations with weight loss in a randomized trial of liraglutide in obese, non-diabetic adults. Int J Obes (Lond) 38(5):689–697. https://doi.org/10.1038/ijo.2013.149
Aronne L, Fujioka K, Aroda V, Chen K, Halseth A, Kesty NC, Burns C, Lush CW, Weyer C (2007) Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo-controlled, dose-escalation study. J Clin Endocrinol Metab 92(8):2977–2983. https://doi.org/10.1210/jc.2006-2003
Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR (2012) The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology 62(5–6):1916–1927. https://doi.org/10.1016/j.neuropharm.2011.12.022
Hjuler ST, Gydesen S, Andreassen KV, Pedersen SL, Hellgren LI, Karsdal MA, Henriksen K (2016) The dual amylin- and calcitonin-receptor agonist KBP-042 increases insulin sensitivity and induces weight loss in rats with obesity. Obesity (Silver Spring) 24(8):1712–1722. https://doi.org/10.1002/oby.21563
Friedman J (2014) 20 years of leptin: leptin at 20: an overview. J Endocrinol 223(1):T1–T8. https://doi.org/10.1530/JOE-14-0405
Ingwersen SH, Khurana M, Madabushi R, Watson E, Jonker DM, Le Thi TD, Jacobsen LV, Tornoe CW (2012) Dosing rationale for liraglutide in type 2 diabetes mellitus: a pharmacometric assessment. J Clin Pharmacol 52(12):1815–1823. https://doi.org/10.1177/0091270011430504
Overgaard RV, Petri KC, Jacobsen LV, Jensen CB (2016) Liraglutide 3.0 mg for weight management: a population pharmacokinetic analysis. Clin Pharmacokinet 55(11):1413–1422. https://doi.org/10.1007/s40262-016-0410-7
Petri KC, Jacobsen LV, Klein DJ (2015) Comparable liraglutide pharmacokinetics in pediatric and adult populations with type 2 diabetes: a population pharmacokinetic analysis. Clin Pharmacokinet 54(6):663–670. https://doi.org/10.1007/s40262-014-0229-z
Hall KD (2008) What is the required energy deficit per unit weight loss? Int J Obes (Lond) 32(3):573–576. https://doi.org/10.1038/sj.ijo.0803720
Gennemark P, Tragardh M, Linden D, Ploj K, Johansson A, Turnbull A, Carlsson B, Antonsson M (2017) Translational modeling to guide study design and dose choice in obesity exemplified by AZD1979, a melanin-concentrating hormone receptor 1 antagonist. CPT 6(7):458–468. https://doi.org/10.1002/psp4.12199
Hall KD, Hammond RA, Rahmandad H (2014) Dynamic interplay among homeostatic, hedonic, and cognitive feedback circuits regulating body weight. Am J Public Health 104(7):1169–1175. https://doi.org/10.2105/AJPH.2014.301931
Acknowledgements
The authors would like to thank Hermann Rapp for valuable discussions and Dr. Arno Kalkuhl for providing us with the toxicology study data. Moreover, we like to thank Sidsel Larsen, Charlotte Holtoft, Arne Lindhardt Jensen, Jessica Bedenik and Christina Doll for excellent technical support.
Author information
Authors and Affiliations
Contributions
All authors provided critical review of drafts of the manuscript, and read and approved the final version. AB and JB contributed equally to the development of the model, to the interpretation of the modeling results and writing the manuscript. All other authors contributed to the design of the study and interpretation of the data, and supported and reviewed the model compilation.
Corresponding author
Ethics declarations
Conflict of interest
Jens M. Borghardt, Wolfgang Rist, Stefan Scheuerer and Tamara Baader-Pagler are employees of Boehringer Ingelheim Pharma GmbH & Co KG. Jolanta Skarbaliene and Maria A. Deryabina are employees of Zealand Pharma A/S. Annika Brings was an employee of Boehringer Ingelheim Pharma GmbH & Co KG at the time of the manuscript creation.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Annika Brings and Jens Markus Borghardt: co-first author.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brings, A., Borghardt, J.M., Skarbaliene, J. et al. Modeling energy intake and body weight effects of a long-acting amylin analogue. J Pharmacokinet Pharmacodyn 45, 215–233 (2018). https://doi.org/10.1007/s10928-017-9557-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-017-9557-6