
Abstract
Influences of methylprednisolone (MPL) and food consumption on body weight (BW), and the effects of MPL on glycemic control including food consumption and the dynamic interactions among glucose, insulin, and free fatty acids (FFA) were evaluated in normal male Wistar rats. Six groups of animals received either saline or MPL via subcutaneous infusions at the rate of 0.03, 0.1, 0.2, 0.3 and 0.4 mg/kg/h for different treatment periods. BW and food consumption were measured twice a week. Plasma concentrations of MPL and corticosterone (CST) were determined at animal sacrifice. Plasma glucose, insulin, and FFA were measured at various times after infusion. Plasma MPL concentrations were simulated by a two-compartment model and used as the driving force in the pharmacodynamic (PD) analysis. All data were modeled using ADAPT 5. The MPL treatments caused reduction of food consumption and body weights in all dosing groups. The steroid also caused changes in plasma glucose, insulin, and FFA concentrations. Hyperinsulinemia was achieved rapidly at the first sampling time of 6 h; significant elevations of FFA were observed in all drug treatment groups; whereas only modest increases in plasma glucose were observed in the low dosing groups (0.03 and 0.1 mg/kg/h). Body weight changes were modeled by dual actions of MPL: inhibition of food consumption and stimulation of weight loss, with food consumption accounting for the input of energy for body weight. Dynamic models of glucose and insulin feedback interactions were extended to capture the major metabolic effects of FFA: stimulation of insulin secretion and inhibition of insulin-stimulated glucose utilization. These models of body weight and glucose regulation adequately captured the experimental data and reflect significant physiological interactions among glucose, insulin, and FFA. These mechanism-based PD models provide further insights into the multi-factor control of this essential metabolic system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucocorticoids (GC) are widely used in a variety of immunosuppressive and anti-inflammatory therapies. Owing to their effects on carbohydrate metabolism, synthetic GC often produce hyperglycemia, insulin resistance, and/or diabetes mellitus. The systemic influences of GC on glucose homeostasis include stimulating hepatic glucose output and inducing insulin resistance in peripheral tissues: two key abnormalities involved in the pathogenesis of steroid diabetes [1]. Numerous studies have examined the mechanisms by which GC alter glucose metabolism and induce insulin resistance in animal models [1–4]. The vital role of GC in regulating glucose homeostasis have been elucidated by either modulating the endogenous steroid [5, 6] or assessing the effects of exogenous GC [2–4], However, the effect of duration of GC treatment on the pathogenesis of metabolic alterations, and the temporal and causal relationships between GC exposure and the extent of glucose/insulin responses have not been well characterized. Clinically, chronic therapy with GC often involves months or even years. The duration of GC exposure is greatly dependent on the appearance of the metabolic side effects and appears to be important in determining the degree of insulin resistance [7].
The GC-induced glucose dysregulation is a whole body metabolic syndrome involving the interplay of multiple target organs. As indicated in a recent PKPD modeling review on diabetes [8], to date most mathematical models focus on glucose and insulin while ignoring most other relevant biomarkers. The role of adipose tissue has become increasingly appreciated and is now recognized as a complex metabolic and endocrine organ [9]. In addition to its function of lipid storage, adipocytes release free fatty acids (FFA) and adipokines which act to communicate with other organs. It has been suggested that GC stimulate lipolysis from adipose tissue, resulting in increased plasma FFA, which serve as an important communicator to other organs and play a critical role in the development of insulin resistance [10]. Treatment with GC elevate circulating FFA in animals [11]. Increased FFA availability and utilization stimulates gluconeogenesis [12–14]. In addition, elevated circulating FFA favor beta-oxidation, resulting in diminished glucose uptake and utilization in peripheral tissues [15]. Moreover, FFA are thought to promote insulin secretion during acute exposure while a long-term excess of FFA lead to impaired pancreatic insulin secretion [10, 16].
Previously, acute bolus and one-week MPL infusion studies in rats were performed to characterize the time course of metabolic alterations in systemic glucose and insulin [17, 18]. The temporary systemic changes after acute dosing were quantified by mechanistic PK/PD modeling. However, the metabolic effects of FFA were not included and the increase in insulin was only accounted for by elevated glucose concentrations. The one-week study may not be long enough to capture all dynamic changes.
In addition to regulating glucose metabolism, GC also modify food intake. The relationship between GC use and change in food consumption has been studied in animal models. Some studies suggested that GC treatment stimulates appetite and increases food intake [19–21], and that this effect might be centrally mediated [19]. In contrast, other studies have reported a reduction in food consumed associated with GC treatment. The reduction in food intake occurring in animals treated with different doses of GC [22–24], has been attributed to induction of leptin expression and reduced production of neuropeptide Y (NPY), a potent stimulator of food intake [22]. The conflicting data indicate that complex factors might be involved in the association between GC and energy intake. Previous studies [17, 18] assumed that the weight loss observed during chronic MPL infusion was mainly catabolic. Therefore, indirect response model IV (stimulation of output) was employed, which did not consider the effect of chronic MPL dosing on food intake. Considering the important role of energy intake in maintaining body weight, we carefully measured body weights and food consumption in this study.
A mechanism-based PD model can provide better quantitative insights into the MPL control of this complex metabolic system. In this report, we investigate food consumption on body weight, explore the influences of FFA on glucose regulation, and develop a model that can capture relationships among GC use and glucose, insulin, and FFA concentrations.
Materials and methods
Animals
Thirty-two male Wistar Rats weighing 310–380 g were purchased from Harlan-Sprague-Dawley Inc. (Indianapolis, IN). All animals were housed under constant temperature (22°C) with free access to water and normal rat chow (Harlan Teklad 2018, Madison, WI), which had a metabolizable energy content of 3.1 kcal/g. Rats were acclimatized to a 12 h/12 h light–dark cycle for at least 1 week before the start of the study. This research adhered to the ‘Principles of Laboratory Animal Care’ and was approved by the University at Buffalo Institutional Animal Care and Use Committee.
Infusion study
Methylprednisolone sodium succinate (Solu-Medrol®, Pharmacia & Upjohn Company, Kalamazoo, MI) was reconstituted with supplied diluent. Rats were randomly divided into 6 sub-groups which received saline infusion (n = 11) or MPL infusions at the rate of 0.03 (n = 4), 0.1 (n = 5), 0.2 (n = 4), 0.3 (n = 3) or 0.4 (n = 5) mg/kg/h via Alzet osmotic mini-pumps (Model 2ML4, flow-rate 2.5 μl/h, DURECT Corp., Cupertino, CA). Rats were sacrificed at various times: 21 days for low dose groups (0.03 and 0.1 mg/kg/h), 10 days for medium dose group (0.2 mg/kg/h), and 7 days for high dose groups (0.3 and 0.4 mg/kg/h). Control rats were also sacrificed at various time points throughout the 21-day period. Complete data could not be obtained from one rat in the control group and one in the 0.1 mg/kg/h dose group, which died before sacrifice. The glucose data from one rat in the medium dose group and insulin data from one rat in the 0.1 mg/kg/h group reached much higher values than others, and were not included in the analysis since inclusion resulted in biased fittings. The MPL concentrations for the pump solution were prepared based on the predose body weight for each rat. After overnight equilibration in saline at 37°C, the pumps were subcutaneously implanted between the shoulder blades at time zero (9:00 a.m.) under phenobarbital anesthesia (50 mg/kg, intraperitoneal injection). All rats were sacrificed by aortic exsanguinations. The body weight of each rat was recorded before pump implantation and daily for the first week and twice a week thereafter. Food intake was measured before pump implantation and twice a week afterwards as the difference between the preweighed amount of rat chow and that remaining. Blood samples were taken from lateral saphenous vein puncture of alternate legs. Blood was drawn into a micro-capillary tubes (44 μl) using EDTA as the anticoagulant. Blood was sampled at predose, 6 h and 1, 2, 3, 4, 5, 6, 7 days for 1 week and twice a week thereafter (10, 14, 17 and 21 days) up to sacrifice time. Blood samples were immediately centrifuged at 2,000×g for 15 min at 4°C, and the plasma samples were frozen at −80°C until analysis.
Assays
Plasma MPL and CST concentrations were quantified simultaneously using a normal-phase high-performance liquid chromatography (HPLC) method with a quantitation limit of 10 ng/ml each [25].
Plasma insulin was measured using the Rat/Mouse Insulin ELISA kit (Millipore Corporation, Billerica, MA). This assay was carried out according to manufacturer’s directions with experimental samples run in triplicate and standards run in duplicate. The CV% were less than 10% and the limit of sensitivity for this assay is 0.2 ng/ml.
Blood glucose was measured using BD Logic Blood Glucose Monitor (BD, Franklin Lakes, NJ), which is a glucose oxidase biosensor. The recommended blood/plasma glucose conversion: Plasma Glucose = 1.11 · Blood Glucose [26] was used.
Plasma free fatty acids were measured using a commercial enzymatic colorimetric assay (Roche Applied Sciences, Indianapolis, IN) adapted to a 96-well plate format. Standards were prepared from a commercial standard solution (WAKO NEFA, WAKO Chemicals, Richmond, VA) with assay linearity range of 0.05–1 mM. This assay involves the formation of hydrogen peroxide (H2O2) by converting FFA to acyl CoA in the presence of acyl CoA synthetase and ATP, followed by subsequent colorimetric detection of H2O2. For the microtiter plate assay, 200 μl Reagent A and 10 μl samples or standards were added to plate wells and incubated for 10 min at room temperature. Thereafter 10 μl Reagent 3 was added to each well to obtain reading 1 (R1), with absorbance read at 546 nm. Then 10 μl Reagent B was added to each well, and after incubation for 60 min at room temperature, the absorbance was measured at 546 nm to obtain R2. FFA concentrations were calculated as R2–R1. Linear regression analysis of all standard curves yielded r2 values of greater than 0.99, and the CV% obtained were less than 15%.
Pharmacokinetic/pharmacodynamic model
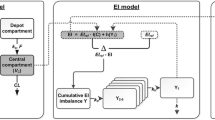
The pharmacokinetics of MPL was described by a two-compartment model with zero-order input into the central compartment (Fig. 1) where MPL p is the plasma methylprednisolone concentration, MPL t is drug in the tissue compartment, CL is drug clearance, V p is central volume of distribution, and k 12 and k 21 are distribution rate constants. The pharmacokinetic parameters were fixed (Table 1) from our previous study [17] and plasma drug concentrations were used as the driving force in the dynamic analysis.

Schematic representation of the PK/PD model of MPL effects on food consumption and body weight. The dotted lines and rectangles indicate inhibition (closed bar) and stimulation (open bar) of the various processes by MPL. Symbols are defined in the text and Table 2
Pharmacodynamic modeling
Food intake and body weight
The model for effects of MPL on food intake and the total body weight of rats is depicted in Fig. 1. An indirect response model was used to describe the daily food consumption (kilocalories per day) with the inhibition function applied to the input rate for food (k in_Food ). The initial conditions of the differential equations for daily food consumption were fixed to the mean baseline values at study day zero for each dose group. Significant reductions in food consumption after MPL treatments were found, hence the drug-specific parameters \( I_{\max }^{Food} \) and \( IC_{50}^{Food} \) were used to represent the maximum inhibition of k in_Food and the plasma MPL concentration required for half-maximal inhibition. We observed an initial decreased food intake in the first week of the study with a gradual increase thereafter in control rats. This may be attributed to a stress effect since blood samples were taken daily for the first week with less frequent blood sampling and animal handling subsequently. The stress effect on food intake was modeled as an exponential decline of k in_Food from k in_Food0 to a new steady-state k in_FoodSS . Further, a feedback step (FB) was needed to model the tolerance effect of the slowly increasing food intake observed in control rats. The ratio of baseline food intake (Food 0) and the daily food consumption (Food) was used to account for the negative feedback, with a first-order rate constant k describing the delayed effect on k in_Food . Similarly, we used indirect response model IV (stimulation of output) to model the catabolic effect of MPL on BW [17]. The pharmacodynamic equations are:
where k in_Food0 = k out_Food · Food max. Parameters k in_Food (kilocalories per day squared) and k out_Food (day−1) are used to describe the input and loss rate for food intake. The Food max indicates hypothetical maximal food consumption at steady-state that would be reached in the absence of MPL and the negative feedback. The initial condition of Eq. 2 is 1, and Food 0 is the initial condition of food intake, which was fixed to the average food consumption at study day zero for each dose group.
Sigmoidal E max functions depict either the inhibitory effect of MPL on the input rate of food intake or the stimulatory effect of MPL on body weight loss. The \( I_{\max }^{Food} \) and \( IC_{50}^{Food} \) represent the maximum inhibition of k in_Food and the plasma drug concentration required at half of \( I_{\max }^{Food} \). The body weight is controlled by the energy input [27] and the first-order loss rate constant \( k_{d}^{BW} \). The efficiency factor (EF) describes the efficiency of converting energy that is needed for metabolism to body weight (grams per kilocalorie). Parameters \( S_{\max }^{BW} \) and \( SC_{50}^{BW} \) describe the maximum stimulation of \( k_{d}^{BW} \) and the plasma MPL concentration required for half-maximal stimulation. The baseline body weight BW 0 was fixed as the mean body weight of animals at study day zero for each dose group. The system is not at steady-state at time zero since rats will keep growing. Once the steady-state body weight has been achieved, the system will produce the relationship: \( EF \cdot Food_{SS} = k_{d}^{BW} \cdot BW_{SS} \), where Food SS and BW SS represent food intake and BW at steady-state in the system.
Glucose–insulin–FFA dynamics
A mechanistic PD model incorporating GC stimulation of glucose and FFA production, glucose–insulin feedback, FFA stimulation of insulin secretion along with an antilipolytic effect of insulin as well as the negative feedback of FFA on insulin stimulation of glucose utilization was developed (Fig. 2). Our previous study in normal rats [17] showed the high potency of MPL for inhibition of CST secretion and low potency of endogenous CST on glucose production, with the potency estimate of 6506 ng/ml which is well above the concentration range in our study (data obtained at animal sacrifice). Therefore, it is reasonable to assume that the major metabolic effects were mediated by MPL. The following equations were used to simultaneously fit glucose, insulin and FFA data from all treatments:
where G, I, and F represent glucose, insulin, and FFA plasma concentrations. Indirect response models were used to describe the turnover of these biomarkers. Glucose is constantly produced with a zero-order rate constant \( k_{s}^{G} \). Considering exogenous glucose input from food intake, a linear efficiency constant S Food is used to drive the food effect on rate of glucose synthesis. A first-order rate constant \( k_{d}^{G} \) describes net removal of glucose by tissue uptake and utilization. MPL acts on glucose presumably via stimulating the production process. The \( S_{\max }^{G} \) describes the maximum achievable stimulation of \( k_{s}^{G} \). The sensitivity parameter \( SC_{50}^{G} \) represents the plasma MPL concentration which produces half-maximal stimulation. Insulin is assumed to control glucose concentration by stimulating its disposition with a linear efficiency constant (S IG ). The change of insulin from its baseline value at time zero (I 0) was used to drive this stimulation effect. The ratio of F 0 /F depicts a negative feedback of FFA on this stimulation effect of insulin on glucose utilization, using a time dependent function to describe the impaired glucose disposition rate under hyperglycemic conditions [17].

In Eq. 6, insulin is produced with a zero-order rate constant \( k_{s}^{I} \) and degraded with a first-order rate constant \( k_{d}^{I} \). The glucose change from its baseline (G 0) stimulates insulin production with a linear efficiency constant S GI . The difference between plasma FFA and its baseline (F 0) stimulates insulin secretion, along with an empirical function exp(−k FI · t) indicating the time-dependent impaired stimulation effect of FFA on insulin secretion. In Eq. 7, FFA is constantly produced with a zero-order rate constant \( k_{s}^{F} \) and utilized with a first-order rate constant \( k_{d}^{F} \). MPL alters FFA production presumably via stimulating lipolysis. \( S_{\max }^{F} \) describes the maximum achievable stimulation of \( k_{s}^{F} \). The sensitivity parameter \( SC_{50}^{F} \) represents the plasma MPL concentration which produces half-maximal stimulation. Insulin is assumed to control FFA concentrations by stimulating disposition. The change of insulin concentration from its baseline at time zero (I 0) was used to drive this stimulation effect with a linear efficiency constant (S IF ). Lower limits of zero were included in Eqs. 5–7 for [1 + S IG · (F 0 /F) · (I − I 0)], [1 + S GI · (G − G 0) + S FI · (F − F 0) · exp(−k FI · t)] and [1 + S IF · (I − I 0)] in order to assure a mathematically plausible relationship in these models.
At time zero, the system was assumed to be at physiological steady-state according to:
where initial values of G 0, I 0, and F 0 were fixed as the mean values from animals at time zero for each dose group. This was necessary to avert overparameterization of this complex system.
Data analysis
Repeated measures analysis of variance was used to test for statistically significant differences in the plasma glucose, insulin, and FFA time courses among treatment groups. This was implemented with the SAS 9.1 procedure MIXED. Differences among treatment means were determined with the least significant difference (LSD) at the 5% significance level. All data are reported as mean ± SD.
The food consumption and body weight data from all dosing groups were fitted simultaneously to Eqs. 1–4, and a piecewise approach was then used for fitting the glucose, insulin and FFA data as parameters were fixed for MPL effects on food consumption. ADAPT 5 software [28] was used for all model fittings with the maximum likelihood method. The variance model was defined as V(σ,θ,t i ) = (σ 1 + Y(θ,t i ) · σ 2)2, where V(σ,θ,t i ) is the variance for the ith point, Y(θ,t i ) is the ith model-predicted value, θ represents the estimated structural parameters, and σ 1 , σ 2 are the variance parameters which were estimated. Proposed PD models for body weight and glucose–insulin–FFA dynamics were fitted and compared. Models were selected based on visual inspection of curve fittings, precision of parameters, Akaike Information Criterion (AIC), Schwarz Criterion (SC) and examination of the residuals.
Results
Pharmacokinetics
The simulated and actual plasma MPL concentration–time profiles are shown in Fig. 3. Due to the short elimination half life (~1 h), infusions quickly produced constant MPL concentrations by the first sampling time point (6 h). Model simulations yielded steady-state concentrations of 6.11, 20.4, 40.7, 61.1, 81.5 ng/ml during 0.03, 0.1, 0.2, 0.3 and 0.4 mg/kg/h MPL infusions, which are in good agreement with the measured plasma MPL concentrations at different sacrifice times.

Steady-state pharmacokinetics of MPL for 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h subcutaneous infusion groups. Symbols are observed drug concentrations. Lines are simulation results using established parameters for 0.03 (light long dash line), 0.1 (heavy long dash line), 0.2 (short dash line), 0.3 (light solid line) and 0.4 (heavy solid line) mg/kg/h infusion groups. The PK parameters are listed in Table 1
Pharmacodynamics
Food intake and body weight
The observed food consumption and BW changes after various doses of MPL are shown in Figs. 4 and 5. In control rats, food consumption was reduced dramatically (about 40%), which may be attributed to the stress of surgery and frequent blood sampling in the first few days [29]. In agreement with this, we observed that food consumption started to increase slowly by day 4. Further, in the control rats, BW initially decreased and then rats slowly regained weight by day 4, which agrees with their pattern of food consumption. The drug caused further reductions of food intake and BW at all dose levels. There were significant dose and time effects on food consumption and BW (P < 0.05). In comparison with the control group, only transitory decreases in food consumption were observed in the low dose groups (0.03 and 0.1 mg/kg/h) and the reductions were not significant. There were significant BW changes in the low dose groups on day 3 and later (P < 0.05). By day 7, there was about 14% BW loss in the 0.03 mg/kg/h dose group and about 21% loss in the 0.1 mg/kg/h group. This is similar to previous results in animals receiving 0.1 mg/kg/h MPL (~19.1%) [17]. In the medium dose group (0.2 mg/kg/h), daily food intake and BW decreased significantly compared to control (P < 0.05). By day 10, about 25% BW loss was observed. Significant reductions were also observed for food intake and BW in the high dose groups (0.3 and 0.4 mg/kg/h). By day 7, there was about 25% BW loss in 0.4 mg/kg/h dose group and 24% loss in 0.3 mg/kg/h dose group, which is consistent with our previous study for rats receiving this dose of MPL (~28%) [17].

The time course of changes in food intake (kilocalories per day) for saline (filled circle), and 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h MPL infusions. Lines depict the simultaneous fitting results for the saline (dotted line), 0.03 (light long dash line), 0.1 (heavy long dash line), 0.2 (short dash line), 0.3 (light solid line) and 0.4 (heavy solid line) mg/kg/h infusion groups with Eqs. 1–4. The PD parameters are listed in Table 2

The time course of changes in body weight (grams per day) for saline (filled circle), and 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h MPL infusions. Lines depict the simultaneous fitting results with Eqs. 1–4. Lines are defined as in Fig. 4. The PD parameters are listed in Table 2
The dynamic model (Fig. 1) well captured the changes of food intake (Fig. 4) and BW (Fig. 5) after various MPL dosing regimens using Eqs. 1–4. The estimated parameters are listed in Table 2. The factor EF depicts the efficiency of converting energy needed for metabolism to body weight and the estimate of 0.510 g/kcal is similar to the value in normal WKY rats (0.302 g/kcal) [27]. The reduction of food consumption in control rats was described by an exponential decay function for the input rate k in_Food for food intake. The estimated rate constant k d of 1.21 day−1 indicates that it will take 2–3 days for the input rate k in_Food to decrease to a new steady-state, which agrees with our experimental observations. The feedback transduction rate k of 0.0531 day−1 indicates a slow adaptation to the reduced food consumption. The \( I_{\max }^{Food} \) of 0.618 suggests that MPL treatment may reduce food consumption to as low as 38.2% of predose intake, assuming there are no stress and adaptive feedback influences. The \( IC_{50}^{Food} \) of 7.32 ng/ml indicates a high potency of MPL to reduce food consumption. The body weight loss rate \( k_{d}^{BW} \) of 0.0765 day−1 is in good accordance with our previous study (0.0792 day−1) [17]. We noticed that in Figs. 4 and 5, BW returns to the basal level at the end of study while food intake still remains much lower than the predose value in control rats. This could be mainly explained by the influences of \( k_{d}^{BW} \) and the dynamic changes of food intake. The smaller \( k_{d}^{BW} \), the larger BW ss that would be achieved and the sooner BW would return to baseline. Our model estimates of Food SS = 65.5 kcal/day and BW SS = 437 g in control rats further confirm the observations. The \( S_{\max }^{BW} \) of 0.118 is lower than found previously (1.04) [17], suggesting that reductions of BW may be related to the ability of GC to act directly or indirectly on various processes. The \( SC_{50}^{BW} \) was fixed to a value of 20.5 ng/ml according to the best model fitting results which is in good agreement with our previous value (16.4 ng/ml) [17].
Glucose–insulin–FFA dynamics
Figures 6, 7 and 8 present the time course of plasma glucose, insulin, and FFA concentrations throughout the study period. A trend of slight decrease in plasma glucose and increase in plasma FFA at early time points was observed in control rats, and a more pronounced decrease in insulin was observed relative to the baseline insulin level in control rats. The drop in plasma glucose concentration could be mainly explained by the reduced food intake in controls, considering the exogenous source of glucose and significant feedback mechanisms among glucose, insulin, and FFA. The decrease in plasma insulin and increase in plasma FFA could be due to the negative effects on insulin production and FFA disposition when plasma glucose and insulin fall below the baseline as indicated in Eqs. 6 and 7. In the low dosing groups, no significant differences in plasma glucose were seen at all time points in comparison with control, although we observed a marginal increase in glucose concentrations. During drug administration, plasma glucose continuously rose from 174 ± 29.3 to 215 ± 25.4 mg/dl in the medium dose group by day 10, from 192 ± 21.4 to 248 ± 33.4 mg/dl with the 0.3 mg/kg/h infusion, and from 211 ± 22.4 to 313 ± 114 mg/dl with the 0.4 mg/kg/h infusion by day 7. Furthermore, the plasma glucose-time profiles for high doses were significantly different from controls (P < 0.05), while the medium dose group only showed a significant difference at the end of study. As shown in Fig. 7, insulin concentrations were markedly increased for all dose groups. For medium and high dose groups, significant differences were seen as early as at the first sampling time point of 6 h (P < 0.05). Insulin profiles showed an early peak at 6 h, declining thereafter but still remaining higher than in control. Dramatic changes were observed in FFA after various MPL treatments (Fig. 8). During drug administration, plasma FFA increased by three- to tenfold starting at the first sampling time of 6 h for all dosing groups, and declined around 1–2 days after treatments.

Plasma glucose versus time profiles during infusions of saline (filled circle), and 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h MPL. Lines depict simultaneous fittings of glucose, insulin, and FFA with Eqs. 5–10. Lines are defined as in Fig. 4. The PD parameters are listed in Table 3

Plasma insulin versus time profiles during infusions of saline (filled circle), and 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h MPL. Lines are simultaneous fittings of insulin concentrations with Eqs. 5–10. Lines are defined as in Fig. 4. The PD parameters are listed in Table 3

Plasma FFA versus time profiles during infusion of saline (filled circle), and 0.03 (open circle), 0.1 (filled inverse triangle), 0.2 (open triangle), 0.3 (filled square) and 0.4 (open square) mg/kg/h MPL. Lines represent simultaneous fitting results with Eqs. 5–10. Lines are defined as in Fig. 4. The PD parameters are listed in Table 3
Model fittings of the plasma glucose, insulin, and FFA concentrations are also shown in Figs. 6, 7 and 8 based on Eqs. 5–10, and the estimated parameters are listed in Table 3. This model represents an extension of previous models incorporating food consumption effects and FFA interactions with glucose and insulin [17]. All data were modeled simultaneously and the fittings adequately captured the time courses of the three biomarkers for various drug doses. In control rats, a slight drop in plasma glucose and insulin, as well as an increase in FFA were captured when the food effect on glucose input was taken into account. Parameter S Food reflects the efficiency of food consumption in controlling the input rate of glucose. The estimate of 0.00832 day/kcal indicates a 100% increase in \( k_{s}^{G} \) when food consumption is 120 kcal/day. Similarly, when food consumption decreases, the exogenous glucose production would decrease. Parameters \( S_{\max }^{G} \), \( SC_{50}^{G} \), and \( S_{\max }^{F} \), \( SC_{50}^{F} \) characterize direct drug effects on glucose and FFA regulation. The estimated \( S_{\max }^{G} \) of 1.14 and \( SC_{50}^{G} \) of 16.6 ng/ml are higher than previous estimates (0.21 and 1.05 ng/ml), indicating that the maximum achievable stimulation of the glucose production rate is 2.14-fold and a relatively lower potency of direct MPL effects on glucose production. Parameters \( S_{\max }^{F} \) of 26.9 and \( SC_{50}^{F} \) of 43.5 ng/ml represent the ability of MPL to achieve maximal stimulation of FFA production as 27.9-fold, indicating that more pronounced FFA changes could be achieved with higher MPL doses.
Parameters \( k_{d}^{G} \), S GI, \( k_{d}^{I} \), S IG , S IF , \( k_{d}^{F} \), and S FI characterize the glucose–insulin–FFA interactions. Rapidly increasing FFA regulates insulin secretion with a simulation efficiency factor S FI of 20.1 mM−1, implying that insulin production would be induced by 100% initially when FFA increase by 0.0497 mM. In addition, the rate constant k FI of 4.41 day−1 suggests that the stimulation effect of FFA on insulin secretion will be diminished within 1 day during drug treatments. Elevated glucose concentrations stimulate insulin secretion, which is represented by the parameter S GI of 0.0178 dl/mg, much lower than a previous estimate of 0.17 dl/mg when the stimulation effect of FFA on insulin secretion was not considered [17]. This indicates that more pronounced changes in glucose are needed to exert the same magnitude increase of insulin production. Elevated insulin secretion from pancreatic β-cells counterbalances the increased plasma glucose with a major effect of increasing peripheral glucose utilization. The ability of insulin to stimulate glucose utilization is represented by the S IG of 0.0399 ml/ng, higher than our previous result 0.011 ml/ng [17], which could be explained by the negative feedback of FFA on this stimulation effect. The antilipolytic effect of insulin is described by a stimulation efficiency factor S IF of 0.315 ml/ng, indicating that the rate of disappearance of plasma FFA would increase 100% when plasma insulin increases by 3.17 ng/ml.
The current glucose dynamic model was chosen based on physiology, previous modeling results [17, 18], and selection after considering alternative model components. These included: direct input of glucose from food into the plasma glucose pool; increased FFA stimulating gluconeogenesis and enhancing glucose production; prolonged hyperglycemia stimulating FFA production from adipose tissue; and relationships between food consumption, glucose, insulin, and FFA. Owing to model complexity, we were not able to incorporate any of these factors. However, the final model structure is consistent with literature results, and the various mechanistic processes included in the model indicate the major interactions among those biomarkers and provide improved insights into GC actions on glucose regulation.
Discussion
Glucocorticoids are key regulators of both stress and energy balance [29]. In this study, reduced food intake in MPL treated rats was observed, in agreement with some literature reports [22–24]. Thus the nutritional influence of food on BW was included in this modeling analysis. Reduction of food consumption observed in the control group might be associated with stress due to pump implantation and frequent blood sampling. Increased CST secretion in response to both acute and chronic stressors can modify feeding behavior in rodents [19]. Elevated CST observed previously indicated such stress effects [17]. The biphasic food consumption profile in control rats suggested that adaptation is occurring which was described by a negative feedback mechanism acting on the food input process. The loss rate for food input to a new steady-state was fixed according to the best fitting results, indicating insufficient information in the first few days to estimate this parameter with good precision. Our estimates for food intake turnover k in_Food0 (171 kcal/day2) were higher than previously reported (~21 kcal/day2) [27]. This could be due to the different strains and considerably lighter animals used previously (~52.3 g), and different experimental design. The relatively low estimate of the hypothetical maximal food intake (54.4 kcal/day) is probably caused by the chronic stress effect during the study. Repeated stressors usually decrease chow intake in rodents [29] inevitably causing changes in body weights. The pronounced BW losses suggest that part of these effects can be ascribed to the increased energy expenditure. In our study, gastrocnemius (GAST) muscles were excised and weighed at animal sacrifice. The GAST muscle weight/BW of individual rats was calculated. Compared to the control group (0.99 ± 0.10%), significant decreases in GAST/BW were observed at all dose groups: 0.84 ± 0.057% for 0.03 mg/kg/h, 0.85 ± 0.20% for 0.1 mg/kg/h, 0.70 ± 0.075% for 0.2 mg/kg/h, 0.69 ± 0.017% for 0.3 mg/kg/h and 0.72 ± 0.10% for 0.4 mg/kg/h. This is consistent with the major catabolic actions of corticosteroids [30] and agrees with our previous findings [31]. Muscle wasting is one of the major adverse effects of chronic steroid therapy and substantial muscle weight loss could be the major cause of BW reduction. The direct catabolic effects of MPL on BW were described by indirect response model IV, with the drug stimulating the degradation rate (Fig. 1). The \( SC_{50}^{BW} \) of 20.6 ng/ml indicates a high potency of MPL in reducing BW. Our model adequately captured both food intake and BW changes in both control and MPL groups (Figs. 4, 5). The estimated parameters for describing BW changes were obtained with good precision and are in good agreement with previous reports [17, 27]. By co-modeling food intake and BW data, we gained better understanding of the quantitative relationship among food intake, body weight, and drug effects.
A physiologic feedback model describing the relationship between glucose and insulin was applied by Lima et al. [32] and often used in the literature [8]. The basic feature is that glucose stimulates production of insulin and insulin stimulates utilization of glucose. The feedback model can be applied to evaluate other drug effects such as describing MPL effects on glucose regulation [17]. Owing to the complex and reciprocal interactions, parameter estimations in such a model can be difficult. Our model was extended based on previous results [17, 18] by adding the food effect and FFA dynamics, and the interactions among various processes (Fig. 2).
It is well known that GC regulate glucose metabolism either by directly affecting glucose production or by opposing the actions of insulin in peripheral tissues by inducing insulin resistance [1–4]. With insulin resistance, the metabolism of proteins, carbohydrates, and lipids are altered [33]. As diabetogenic agents, the risk of developing or aggravating diabetes increases as GC doses or duration of treatment increase. In our study, animals treated with low doses of MPL only showed marginal elevated glucose concentrations. However, the high dosing groups exhibited significantly higher glucose concentrations throughout most of the study period, and dose-dependent drug effects on glucose regulation were observed. As glucose homeostasis is maintained via functions of β-cells, liver, muscle and adipose tissue, numerous factors could be involved in the adaptive responses during GC treatments. Among these, β-cell function appears to be a major contributor since hyperinsulinemia often accompanies and could counteract the GC-induced glucose disturbances. Thus, owing to the antagonistic effects of insulin and the large functional reserve of β-cells, GC-induced hyperglycemia could be compensated by appropriate elevations of insulin secretion. Consistent with this view, GC-treated animals are often hyperinsulinemic [4, 22]. During prolonged GC treatments, animals exhibit increased β-cell mass as well as degranulation of pancreatic β-cells [2]. Depending on the dose and time of GC treatments, different degrees of insulin resistance can occur [4]. Although moderate hyperinsulinemia is capable of preventing any increases in plasma glucose after relatively low doses of GC treatments [4, 22], significant higher insulin concentrations may not be sufficient to maintain normal glucose with higher doses of GC [4]. Therefore, proper coregulation of insulin sensitivity and insulin secretion represent the key features of glucose homeostasis. With inappropriate metabolic adaptations, compensatory failure results in full blown “steroid diabetes”. Other possible explanations for such adaptations include modified insulin action via changing insulin binding [1] and alterations in GC-induced insulin resistance such as down-regulation of glucocorticoid receptors [3] or altered plasma FFA [10, 16]. Among these, the role of circulating FFA has gained particular attention.
As increased circulating FFA are often accompanied by hyperinsulinemia in GC-treated animals [4, 22], evidence suggests that GC promote lipolysis by the activation of hormone-sensitive lipase [34] and inhibition of lipoprotein lipase [35], thus resulting in elevated blood FFA. In our study, this is also reflected by the fact that various MPL doses produced marked elevations in circulating FFA. Significant increases in plasma FFA were found very early and these elevations are consistent with results from dexamethasone-treated rats [22]. In addition, the estimated maximal stimulation factors of \( S_{\max }^{F} \) (26.9) and \( S_{\max }^{G} \) (1.14) indicate the greater potential of MPL to stimulate changes in FFA than of glucose.
The role of FFA on glucose regulation and insulin resistance has become increasingly appreciated [9, 36]. The FFA play a critical role in modulating β-cell function and insulin secretion [37], and are also important in mediating the compensatory responses to GC-induced insulin resistance [22, 36]. Acute elevations of plasma FFA stimulate insulin secretion [38, 39], whereas chronic effects of FFA on insulin secretion were reported with less clear results [36]. In vitro studies using isolated rat islets [40] or perfused rat pancreas [41] showed biphasic insulin response profiles with FFA administration: initial near-maximal increase at 6 h followed by a decrease in insulin secretion, with about 40% reduction in insulin content in fatty acid-exposed rat islets after 6 h [40]. This is consistent with our observations of biphasic patterns of plasma insulin (Fig. 7). In our dynamic model, it was estimated that a 100% increase in insulin production would occur with a 0.049 mM increase in plasma FFA, corresponding to about a 100% increase from the basal FFA level. In the literature, about a 150% increase in plasma insulin was reported when endogenous plasma FFA was elevated by 100% [42], close to our predictions. An empirical function exp(−k FI · t) was used to describe the time-dependent stimulation action of FFA on insulin secretion. The estimated rate constant k FI of 4.41 day−1 and the high potential of FFA to stimulate insulin secretion suggest that the initial spike of insulin may be driven by the increased circulating FFA. On the other hand, animal studies also indicate that markedly elevated FFA (> 1.5 mM) could be toxic to β-cells and impair insulin secretion, according to a lipotoxicity hypothesis proposed by Unger [43]. In our study, the highest observed plasma FFA was 0.68 ± 0.29 mM, thus the negative effect of FFA on insulin secretion might not be relevant.
The chronically elevated circulating FFA, although counteracting insulin resistance by enhancing insulin secretion from β-cells, could in parallel impair peripheral insulin sensitivity. Increased concentrations of FFA promote insulin resistance by inhibiting insulin-stimulated glucose uptake and glycogen synthesis in muscle and also inhibit insulin-mediated suppression of glycogenolysis in liver [10, 15, 44]. Preferential oxidation of FFA over glucose, defects in insulin signaling, and impaired glucose transport have been suggested to be responsible for insulin resistance [15]. The fact that many obese people with elevated plasma FFA do not develop diabetes is intriguing [44]. FFA-mediated hyperinsulinemia was proposed to compensate for FFA-induced insulin resistance [22, 36], and sufficient insulin secretion, together with appropriate β-cell function are important in maintaining glucose homeostasis. Similarly, it was suggested that elevations in circulating FFA after GC treatment play an important role in both adaptive glucose regulation and insulin resistance. A negative feedback of FFA on the stimulation efficiency of insulin on glucose utilization was incorporated into the dynamic model, indicating the other key abnormality of glucose regulation in GC-treated animals. In the model, time course changes of F 0 /F were used to modify the stimulation efficacy factor S IG . As FFA increase, the ability of insulin to promote glucose utilization decreases, leading to the development of insulin resistance.
In addition to serving as an important regulator of glucose metabolism, insulin plays a major role in lipid metabolism: insulin stimulates utilization of plasma FFA by promoting triglyceride synthesis in adipose tissue. A comprehensive dynamic model was developed recently [45] which recognized the significant interactions among glucose, insulin, and FFA and extended the original minimal model [46] to include FFA dynamics. Three major metabolic effects were described and captured in this model: glucose stimulating FFA production, insulin promoting FFA utilization, and FFA impairing insulin action on glucose uptake. Due to the complexity in the model structure, piecewise fitting was applied during a euglycemic clamp study [47]. However, similar to the minimal model, the simultaneous interactions between glucose and insulin were not taken into account and some curve fittings were not shown. In our model, including glucose stimulation of FFA production generated poor parameter estimates and fittings. In addition to the effects of FFA on peripheral insulin actions, the antilipolytic effect of insulin on FFA was modeled using a linear stimulation coefficient S IF to describe the efficiency of change in plasma insulin in stimulating FFA utilization. Our model estimate of S IF = 0.315 ml/ng, corresponds to a maximal 50% decrease in FFA when plasma insulin increases to 3.17 ng/ml, assuming no influence of drug effects. The published plasma FFA profiles showed similar patterns during an intraperitoneal insulin tolerance test in DEX-treated and control animals [22]. A maximum 50% decrease in plasma FFA observed in both groups was seen with insulin administration, although there were about threefold differences in initial insulin concentrations in these two groups.
Various indirect mechanisms, multiple component interactions, and variability in experimental data posed challenges in our model fittings. Some parameters (\( k_{d}^{F} \), S IG and \( k_{d}^{I} \)) were fixed to the best current fitting results. These parameter values are either similar to previous results in our lab [17] or close to literature reported values of FFA turnover [48–50]. The average estimates for the k out parameter for loss of FFA from plasma range from 0.06 to 0.30 min−1 [48–50], close to our \( k_{d}^{F} \) value (225 day−1 or 0.16 min−1). Assuming that plasma glucose is 200 mg/dl and baseline food intake is 100 kcal/day, the calculated glucose production rate \( k_{s}^{G} \) · (1 + S Food · Food 0) is 0.096 mg/dl/min. This is near the literature value of 1 mg/kg/min for hepatic glucose output [51], assuming the volume of distribution of glucose in rats is 2.9 dl/kg [52].
Some limitations of the proposed dynamic models exist. Appreciable variability was seen in the various biomarker responses (Figs. 6, 7, 8). This may be attributed to different sensitivities of individual rats to MPL and to animal handing and stress. Additional interactions among glucose, insulin, and FFA involved in glucose regulation were left out of the final model to avoid overparameterization. Many pathophysiological effects of GC are mediated by their transcriptional regulation of target genes, with these effects primarily dependent on the interaction with their receptors (GR) [53]. Extension of the current model could be achieved by measuring receptor binding components and target gene mRNA in different organs to fully account for the receptor/gene-mediated GC effects. Our previous study measured CST in normal rats and incorporated the circadian rhythm of CST into the final model in order to differentiate endogenous and exogenous GC effects [17]. From the measured CST at animal sacrifice (data not shown), MPL fully suppressed CST secretion in the 0.1, 0.2, 0.3 and 0.4 mg/kg/h infusion groups. Similar CST (within 100–600 ng/ml) were obtained at sacrifice in control and 0.03 mg/kg/h dose groups which are well below the previously estimated potency of endogenous CST on glucose regulation (6506 ng/ml) [17]. Thus, effects of endogenous CST on glucose and body weights are likely negligible compared to exogenous MPL exposures. However, higher CST are expected in control rats due to stress. Finally, multiple organs are involved in GC-mediated glucose disturbances and the current model only included several systemic biomarkers.
In summary, dynamic models were developed to describe MPL effects on body weight and glucose regulation in rats. We integrated food intake and body weights in a mechanism-based model. The steroid infusions caused marked reductions in food intake and body weights, and joint modeling help differentiate direct and indirect MPL actions on body weight. The classic glucose–insulin feedback model was extended by incorporating food and FFA effects. Appreciable changes in plasma FFA during MPL infusions reflect a critical role in regulating adaptive glucose responses and the development of insulin resistance. Integrated modeling of the physiological interactions among glucose, insulin, and FFA suggested the important roles of these entities in both normal β-cell function and insulin sensitivity.
References
McMahon M, Gerich J, Rizza R (1988) Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev 4:17–30
Ogawa A, Johnson JH, Ohneda M, McAllister CT, Inman L, Alam T, Unger RH (1992) Roles of insulin resistance and beta-cell dysfunction in dexamethasone-induced diabetes. J Clin Invest 90:497–504
Stojanovska L, Rosella G, Proietto J (1990) Evolution of dexamethasone-induced insulin resistance in rats. Am J Physiol 258:E748–E756
Rafacho A, Giozzet VA, Boschero AC, Bosqueiro JR (2008) Functional alterations in endocrine pancreas of rats with different degrees of dexamethasone-induced insulin resistance. Pancreas 36:284–293
Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G (2004) Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol 18:1346–1353
Gettys TW, Watson PM, Taylor IL, Collins S (1997) RU-486 (Mifepristone) ameliorates diabetes but does not correct deficient beta-adrenergic signalling in adipocytes from mature C57BL/6J-ob/ob mice. Int J Obes Relat Metab Disord 21:865–873
Selyatitskaya VG, Kuz’minova OI, Odintsov SV (2002) Development of insulin resistance in experimental animals during long-term glucocorticoid treatment. Bull Exp Biol Med 133:339–341
Landersdorfer CB, Jusko WJ (2008) Pharmacokinetic/pharmacodynamic modelling in diabetes mellitus. Clin Pharmacokinet 47:417–448
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Mlinar B, Marc J, Janez A, Pfeifer M (2007) Molecular mechanisms of insulin resistance and associated diseases. Clin Chim Acta 375:20–35
Buren J, Lai YC, Lundgren M, Eriksson JW, Jensen J (2008) Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch Biochem Biophys 474:91–101
Chu CA, Sherck SM, Igawa K, Sindelar DK, Neal DW, Emshwiller M, Cherrington AD (2002) Effects of free fatty acids on hepatic glycogenolysis and gluconeogenesis in conscious dogs. Am J Physiol Endocrinol Metab 282:E402–E411
Chen X, Iqbal N, Boden G (1999) The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest 103:365–372
Williamson JR, Kreisberg RA, Felts PW (1966) Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc Natl Acad Sci USA 56:247–254
Delarue J, Magnan C (2007) Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care 10:142–148
Nicod N, Giusti V, Besse C, Tappy L (2003) Metabolic adaptations to dexamethasone-induced insulin resistance in healthy volunteers. Obes Res 11:625–631
Jin JY, Jusko WJ (2009) Pharmacodynamics of glucose regulation by methylprednisolone. II. Normal rats. Biopharm Drug Dispos 30:35–48
Jin JY, Jusko WJ (2009) Pharmacodynamics of glucose regulation by methylprednisolone. I. Adrenalectomized rats. Biopharm Drug Dispos 30:21–34
Dallman MF, la Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF (2004) Minireview: glucocorticoids—food intake, abdominal obesity, and wealthy nations in 2004. Endocrinology 145:2633–2638
Okada S, York DA, Bray GA (1992) Mifepristone (RU 486), a blocker of type II glucocorticoid and progestin receptors, reverses a dietary form of obesity. Am J Physiol 262:R1106–R1110
Langley SC, York DA (1990) Effects of antiglucocorticoid RU 486 on development of obesity in obese fa/fa Zucker rats. Am J Physiol 259:R539–R544
Novelli M, Pocai A, Chiellini C, Maffei M, Masiello P (2008) Free fatty acids as mediators of adaptive compensatory responses to insulin resistance in dexamethasone-treated rats. Diabetes Metab Res Rev 24:155–164
Barbera M, Fierabracci V, Novelli M, Bombara M, Masiello P, Bergamini E, De Tata V (2001) Dexamethasone-induced insulin resistance and pancreatic adaptive response in aging rats are not modified by oral vanadyl sulfate treatment. Eur J Endocrinol 145:799–806
De Vos P, Saladin R, Auwerx J, Staels B (1995) Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J Biol Chem 270:15958–15961
Haughey DB, Jusko WJ (1988) Analysis of methylprednisolone, methylprednisone and corticosterone for assessment of methylprednisolone disposition in the rat. J Chromatogr 430:241–248
D’Orazio P, Burnett RW, Fogh-Andersen N, Jacobs E, Kuwa K, Kulpmann WR, Larsson L, Lewenstam A, Maas AH, Mager G, Naskalski JW, Okorodudu AO (2005) Approved IFCC recommendation on reporting results for blood glucose (abbreviated). Clin Chem 51:1573–1576
Landersdorfer CB, DuBois DC, Almon RR, Jusko WJ (2009) Mechanism-based modeling of nutritional and leptin influences on growth in normal and type 2 diabetic rats. J Pharmacol Exp Ther 328:644–651
D’Argenio DZ, Schumitzky A, Wang X (2009) ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. BioMedical Simulations Resource, Los Angeles
Dallman M, Bhatnagar S (2001) Chronic stress and energy balance: role of the hypothalamo/pituitary/adrenal axis. In: McEwen BS (ed) Handbook of physiology, section 7: the endocrine system, vol VII. Oxford University Press, New York, pp 179–210
Devenport L, Knehans A, Sundstrom A, Thomas T (1989) Corticosterone’s dual metabolic actions. Life Sci 45:1389–1396
Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ (2002) Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn 29:1–24
Lima JJ, Matsushima N, Kissoon N, Wang J, Sylvester JE, Jusko WJ (2004) Modeling the metabolic effects of terbutaline in beta2-adrenergic receptor diplotypes. Clin Pharmacol Ther 76:27–37
Qi D, Rodrigues B (2007) Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am J Physiol Endocrinol Metab 292:E654–E667
Slavin BG, Ong JM, Kern PA (1994) Hormonal regulation of hormone-sensitive lipase activity and mRNA levels in isolated rat adipocytes. J Lipid Res 35:1535–1541
Ong JM, Simsolo RB, Saffari B, Kern PA (1992) The regulation of lipoprotein lipase gene expression by dexamethasone in isolated rat adipocytes. Endocrinology 130:2310–2316
Boden G (2005) Free fatty acids and insulin secretion in humans. Curr Diab Rep 5:167–170
Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M (2006) Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55(Suppl 2):S16–S23
Pelkonen R, Miettinen TA, Taskinen MR, Nikkila EA (1968) Effect of acute elevation of plasma glycerol, triglyceride and FFA levels on glucose utilization and plasma insulin. Diabetes 17:76–82
Crespin SR, Greenough WB III, Steinberg D (1973) Stimulation of insulin secretion by long-chain free fatty acids. A direct pancreatic effect. J Clin Invest 52:1979–1984
Zhou YP, Grill VE (1994) Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 93:870–876
Sako Y, Grill VE (1990) A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology 127:1580–1589
Stein DT, Esser V, Stevenson BE, Lane KE, Whiteside JH, Daniels MB, Chen S, McGarry JD (1996) Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest 97:2728–2735
Unger RH (1995) Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 44:863–870
Boden G (2001) Free fatty acids-the link between obesity and insulin resistance. Endocr Pract 7:44–51
Roy A, Parker RS (2006) Dynamic modeling of free fatty acid, glucose, and insulin: an extended “minimal model”. Diabetes Technol Ther 8:617–626
Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68:1456–1467
Howard BV, Klimes I, Vasquez B, Brady D, Nagulesparan M, Unger RH (1984) The antilipolytic action of insulin in obese subjects with resistance to its glucoregulatory action. J Clin Endocrinol Metab 58:544–548
Isaksson C, Gabrielsson J, Wallenius K, Peletier LA, Toreson H (2009) Turnover modeling of non-esterified fatty acids in rats after multiple intravenous infusions of nicotinic acid. Dose Response 7:247–269
van Schaick EA, de Greef HJ, Ijzerman AP, Danhof M (1997) Physiological indirect effect modeling of the antilipolytic effects of adenosine A1-receptor agonists. J Pharmacokinet Biopharm 25:673–694
Van der Graaf PH, Van Schaick EA, Visser SA, De Greef HJ, Ijzerman AP, Danhof M (1999) Mechanism-based pharmacokinetic-pharmacodynamic modeling of antilipolytic effects of adenosine A(1) receptor agonists in rats: prediction of tissue-dependent efficacy in vivo. J Pharmacol Exp Ther 290:702–709
Nawano M, Oku A, Ueta K, Umebayashi I, Ishirahara T, Arakawa K, Saito A, Anai M, Kikuchi M, Asano T (2000) Hyperglycemia contributes insulin resistance in hepatic and adipose tissue but not skeletal muscle of ZDF rats. Am J Physiol Endocrinol Metab 278:E535–E543
Sato S, Katayama K, Kakemi M, Koizumi T (1988) A kinetic study of chlorpromazine on the hyperglycemic response in rats. II. Effect of chlorpromazine on plasma glucose. J Pharmacobiodyn 11:492–503
Hayashi R, Wada H, Ito K, Adcock IM (2004) Effects of glucocorticoids on gene transcription. Eur J Pharmacol 500:51–62
Acknowledgments
Financial support for this research was provided by Grant GM24211 from the National Institute of Health. The authors thank Ms. Nancy Pyszczynski for performing the HPLC assay.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fang, J., DuBois, D.C., He, Y. et al. Dynamic modeling of methylprednisolone effects on body weight and glucose regulation in rats. J Pharmacokinet Pharmacodyn 38, 293–316 (2011). https://doi.org/10.1007/s10928-011-9194-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-011-9194-4