
Abstract
In this study, a new perovskite solid solution system of La1−xNaxCrO3 (0 ≤ x ≤ 0.15) was synthesized by sol–gel method using citric acid as chelating agent. The samples were subjected to various calcination temperatures in order to investigate the physicochemical properties of the oxide affected by the parameter. Thermogravimetric analysis, Fourier transform infrared spectroscopy and X-ray diffraction (XRD) techniques are used to explore precursor decomposition and to establish adequate calcination temperature for the preparation of the nanopowders. A high crystallized La0.9Na0.1CrO3 with a orthorhombic structure was obtained when the precursor was calcined at 700 °C in air for 6 h. The studied compounds obtained after calcination at 700 °C were characterized by XRD, Brunauer–Emmett–Teller surface area analysis, scanning electron microscopy, powder size distribution and electrochemical measurements. The microstructure and morphology of the compounds show that the particles are nearly spherical in shape and are partially agglomerated. While the crystallite size decreases with increasing sodium content. The highest surface is achieved for La0.9Na0.1CrO3 oxide. The electrochemical measurements indicate that the catalytic activity is strongly influenced by sodium doping. The highest electrode performance is achieved with large sodium content.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In recent years, there has been a continuous interest in developing new materials with perovskite structure. Perovskite oxides have the general formula ABO3, where the B-site cation occupies the interstitial site of an octahedron of oxygen anions and the A-site cation fits in the large cavity at the centre of the 12 coordination site [1, 2]. Showing a wide range of oxygen stoichiometry, these materials have been attracting much attention due to the existence of mixed ionic and electronic conductivity, and find application in several fields such as catalysis, fuel cells, oxygen sensors, and membranes for gas separations [3, 4]. It has been demonstrated that the existence of mixed valence states of the B ion plays an essential role in determining the physical properties of this kind of materials [5].
The lanthanum chromite perovskite LaCrO3 and compositions derived from it by substituting lanthanum by other metal cations (La1−xMxCrO3) present a high mechanical and chemical stability while displaying high melting points (~2500), large electrical conductivity and a relatively high catalytic oxidation activity [5,6,7,8]. It has been reported in the literature [9,10,11,12,13] that lanthanum chromite, doped with alkaline earth substituent, has high p-type conductivity and are found be stable over vide range of oxygen partial pressure. Due to its stability in low partial pressure, LaCrO3 based materials also find application as interconnects for SOFC [14, 15].
Doped lanthanum chromites have been synthesised by various techniques such as sol gel [16], freeze-drying [17], citrate gel combustion [18] and solid state routes [19]. Among them, high surface area along with high chemical homogeneity at structural level can in principle be achieved by the freeze-drying and sol–gel amorphous citrate methods [17]. Electrical properties of this system have been studied extensively and reported in the literature in pure and doped forms. Sr doped lanthanum chromite, La1−xSrxCrO3, showed thermally activated temperature dependence of electrical conductivity over a vast temperature range [11]. This system was found to display temperature dependent carrier mobility rather than carrier concentration. The La0.7Sr0.3Mn1−xCrxO3 system is reported to exhibit a change from semi-insulating to metallic behaviour at high concentration of Cr [20]. Liu et al. [21] have also reported the effect of Cr6+ ions on the electrical conductivity of La1−xSrxCrO3. Ca doped LaCrO3 was found to show thermally activated small hole-polaron hopping conduction and was found to depict decreasing electrical conductivity with increasing concentration of Ca [22]. However, Jiang et al. [23] have reported that for La1−xMxCrO3 (with M = Mg, Ca, Ba, Sr), Ca and Sr doped compositions exhibited highest conductivities. It has been reported that Ca doped lanthanum chromite and Gd doped ceria composite can potentially serve as an electrode for symmetric SOFC operation [24]. Ong and Wu [25] have also reported that electrical conductivity of lanthanum chromite increased by doping of Ca and Ba. The effect was found to be more pronounced for the Ba doped system. Zhang et al. [16] have measured electrical conductivity of mixed doped La0.9Ca0.1Cr0.5Ni0.5O3 system and showed a phase transition at 192 °C. The doping of divalent elements (like Ca, Sr, and Ba) at trivalent sites (La site) in lanthanum chromite produced more holes in the valence band, which reduced the energy band gap resulting into an increase in conductivity of this system [26]. The heterovalent substitution in the lanthanum chromite system has been studied by various research groups, however, there are only a few studies reported on the isovalent substituted lanthanum chromite system.
To our best knowledge, La1−xNaxCrO3 is a novel oxide, compositions that have not been reported previously. In our present work, samples of the La1−xNaxCrO3 (0 ≤ x ≤ 0.15) were synthesized in order to evaluate the influence of the amount of sodium on the thermal and structural properties of the system. The synthesis was carried out by the citric acid sol–gel method using absolute ethanol as solvent, and characterized by thermogravimetric analysis (TGA), infrared spectroscopy (IR), X-ray diffraction (XRD), Brunauer–Emmett–Teller specific surface area (SBET) analysis, scanning electron microscopy (SEM), powder size distribution (PSD), and cyclic voltametry.
2 Experimental Procedure
2.1 Synthesis of La1−xNaxCrO3 Perovskites
La1−xNaxCrO3 oxides with (0 ≤ x ≤ 0.15) were prepared by a sol–gel method using citric acid as a complexing agent. Aqueous solutions of La(NO3)3·6H2O (BIOCHEM), Na(NO3) (BIOCHEM), and Cr(NO3)3·9H2O (BIOCHEM) were first dissolved in absolute ethanol separately. The molar amount of citric acid was equal to total molar amount of metal nitrates in solution. Citric acid solution was added to the precursors under vigorous stirring. The homogeneous solution obtained was heated at 80 °C for 4 h. To remove the solvent, the gel is heated for 24 h at 100 °C. The resulting precursor is then milled and calcined in air for 6 h at different calcination temperatures: 400, 500, 600, 700, 800 and 1000 °C with the heating rate of 5 °C min−1.
2.2 Characterization Techniques
The thermal decomposition processes of the precursor gels were studied in air atmosphere by TGA using a LINSEIS STA PT1600 with a heating rate of 10 °C min−1 in air, from room temperature to 900 °C. Infrared transmission spectra were performed on a Fourier transform spectrometer (FTIR) Shimadzu 8400 S. A granular technique employing KBr (1 mg of sample added to 200 mg of KBr) was used, and the spectra were recorded in the 400–4000 cm−1 range.
XRD patterns were collected on a Bruker AXS D 8 advance diffractometer employing Cu Kα radiation. In all diffractograms, a step size of 0.02° (2θ) was used with a data collection time of 15 s. Data were collected between 2θ values of 10° and 80° using standard θ/2θ geometry. Identification of crystalline phases was carried out by comparison with JCPDS standards. The unit cell parameters were obtained by fitting the peak position of the XRD pattern using the CELREF and X’pert Highscore programs. Moreover, morphological aspects of the powders were examined by using an ESEM-FEI Quanta 600 FEG scanning electron microscope.
The analysis of the distribution of the grain size of the samples was employed in order to show the influence of the La/Na substitution and the synthetic method employed on the particle size by laser granulometry. After calcination at 700 °C, the powder was dispersed in deionized water in a beaker with magnetic stirring and combined under ultrasound for 15 min. Powder size distribution was characterized with a laser particle size analyzer (Mastersizer 2000, Malvern).
The specific surface area of the samples (SBET) was determined by applying the BET method to nitrogen adsorption/desorption isotherms recorded at −196 °C, using a micrometrics apparatus model Nova series 2000 Quantachrome automatic sorptiometer. Prior to adsorption, the samples were degassed overnight at 150 °C. Pore volume (single point adsorption total pore volume of pores less than ca. 80 nm diameter at P/P0 ca. 0.98 and t-plot micropore volume) was also determined from corresponding analysis of the isotherms.
The electrochemical experiments for O2 reduction and evolution were performed using a Volta Lab 40 potentiostat/galvanostat. The measurements were carried out in a three-compartment cell. Potassium hydroxide electrolyte solution (1 M) was prepared by dissolving the required amount of KOH (Merck) into bidistilled water. The working electrodes (1 cm2) were obtained by painting with an oxide suspension. The counter electrode used was a Pt plate. The reference electrode was Hg/HgO/1 M KOH. All potentials in the text have been referred to this reference electrode.
3 Results and Discussion
3.1 Thermogravimetric Analysis
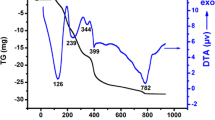
The precursor La0.9Na0.1CrO3 (after the drying step of the preparation at 100 °C) was examined by TGA/DTG in order to explore its decomposition under atmospheric air and with the aim of establishing most adequate calcination conditions. The results are shown in Fig. 1. Basically, four decomposition processes are identified. In the first temperature region, a relatively small weight loss (of ca. 7.64%) is observed between 25 and ca. 170 °C (with an inflection point at ca. 100 °C), This most likely corresponds mainly to desorption of chemisorbed or hydration water that may remain in the precursor, in accordance with previous works [27,28,29]. The second one represents an important mass loss (ca. 21.03%) and takes place between ca. 170 and 340 °C, Such mass loss appears consistent with the oxidative decomposition of citrates complexing the metals in the precursors [30, 31]. The third one takes place between ca. 340 and 560 °C and corresponds to a mass loss of about (ca. 21.73%). It could be related to the decomposition of the remaining carbonate and carboxylate-type complexes in the samples to produce an intermediate chromate phase La0.9Sr0.1CrO4 solid solution [32, 33], in accordance with XRD results presented below. A final weight loss (ca. 9.94%) is observed above 560 °C. This process most likely corresponds mainly to an oxygen loss from reduction of an intermediate lanthanum chromate phase resulting in final generation of the lanthanum chromite perovskite, in agreement with FTIR and XRD results discussed below.

Thermogravimetric profiles of La0.9Na0.1CrO3 precursor powders with a heating rate of 10 °C/min
3.2 IR Spectroscopy
The vibration frequencies in the infrared region are fundamental for the control of the reaction process and properties of materials. Fourier transformed infrared spectroscopy results of La0.9Na0.1CrO3 precursor calcined at different temperatures are shown in Fig. 2. From the spectra of the sample calcined at 400 °C, several absorption bands are observed at about 710, 830, 1250–1650 and 3410 cm−1. The broad absorption bands appear at 1250–1650 cm−1 can probably be ascribed to the stretching vibrations of carbonate group and nitrate anions [34,35,36]. The intensity of these bands decreases after the heating and they become less visible at high temperatures, suggesting the decomposition of the residual material of the synthesis process. These results are in agreement with the TG/DTG analysis. The bands which appear at 710 and 830 cm−1, according to the XRD results, it can be related to the secondary phase (La0.9Na0.1CrO4). The intensity of the bands at 710 and 830 cm−1 decrease significantly after heating at 500 °C, and disappear at higher temperatures (T > 700 °C). These bands must be related to structural vibrations of an intermediate oxide formed at lower temperature than the La0.9Na0.1CrO3 perovskite (see XRD section).

Infrared spectra of La0.9Na0.1CrO3 powder sample calcined at different temperatures
The spectra of samples calcined at 500–800 °C show that two new peaks appear in the region of ~400–700 cm−1, typical of perovskite oxides [37]. Typically, the M–O (M=Cr) vibrations of octahedral MO6 units dominate the spectra [37]. The bands at higher wavenumbers (620–650 cm−1) are assigned to the stretching modes of the MO6 octahedral unit and those at 400–440 cm−1 to the deformation of this same polyhedral unit [37, 38]. Note that a small band at ca. 3410 cm−1 typical of the presence of some water in the KBr employed to dilute the samples into the pellets and/or chemisorbed from the atmosphere onto the samples is also observed in some of the spectra.
3.3 X-ray Diffraction
The powder of La0.9Na0.1CrO3 oxide calcined at different temperatures was analyzed by XRD (Fig. 3). All diffractograms relating to these systems have been recorded in the same operating conditions. From Fig. 3, the precursor calcined at 400 °C for 6 h shows a crystalline nature with too many short and blunt peaks corresponding to the majority impurity phases of La0.9Na0.1CrO4 and minor peaks of Cr2O3. All X-ray diffraction lines (marked with asterisk Fig. 3) for this sample are similar to those of LaCrO4, [39] and could be indexed as a monoclinic unit cell with a = 7.032, b = 7.215, c = 6.671 Å, and β = 104.53°, agreeing well with crystallographic data a = 7.041, b = 7.237, c = 6.693 Å, and β = 104.94° (JCPDS 00-049-1710). The small differences found in the lattice parameters are due to the fact that the present compound is not a pure lanthanum chromate but a chromate phase solid solution.

XRD patterns of the La0.9Na0.1CrO3 powder calcined at different temperatures: (*) La0.9Na0.1CrO4, (+) Cr2O3
When the precursor was heated over 400 °C in air for 6 h, the characteristic diffraction peaks of La0.9Na0.1CrO4 decrease strongly and disappear at 700 °C, while those of orthorhombic perovskite structure appear (JCPDS 00-024-1016). At 700 °C the intensity of characteristic diffraction peaks of orthorhombic perovskite becomes stronger and no reflections from secondary phase are observed as distinct intermediate phases to the formation of La0.9Na0.1CrO3 during the thermal decomposition of the precursor powder. Moreover, these results reconfirm also that the a weight loss of ca. 9,94% in the TGA Fig. 1 above 550 °C is due to the transformation of the byproduct into the final perovskite-type phase by the simultaneous release of O2 as follows : La0.9Na0.1CrO4→La0.9Na0.1CrO3 + ½O2.
Additionally, the purity of the orthorhombic chromite phase was preserved even after thermal treatment at 1000 °C, suggesting that the compound remains stable even at elevated temperatures.
On the basis of these results, and in agreement also with TG/DTG and FT-IR analysis, 700 °C has been chosen as the most adequate calcination temperature to obtain by this method optimum balance between achievement of the chromite perovskite structure and maintenance of the largest possible surface area. It must be noted that this latter can appreciably decrease as a consequence of thermal sintering if higher calcination temperature is applied [32].
Figure 4 shows the XRD patterns of the La1−xNaxCrO3 (0 ≤ x ≤ 0.15), sintered at 700 °C for 6 h in air. The samples with 0 ≤ x ≤ 0.10 were found to be single phase and all the reflections were indexed on the basis of orthorhombic perovskite structure (JCPDS 00-024-1016), with no detectable secondary phase. With the addition of 15% of Na into LaCrO3, it was found that a second phase was observed. The extra reflections from the second phase are consistent with the reflections of Na2CrO4 (JCPDS 00-001-0927). This result indicates that the solubility of sodium cations in LaCrO3 perovskite lattice is less than 15%. This low solubility is probably due to the difference in the ionic radius between Na1+ and La3+ (\({r}_{{{Na}}}^{{1+}}\) = 1.39 Å, \({r}_{{{La}}}^{{3+}}\) = 1.36 Å) [40]. This is in good agreement with a previous work on (La1−xA) AlO3 (A = Ca, Sr, Ba) [41] which indicates that the radius of cation play an important role on the dissolution behavior of A-site-doped LaAlO3. In addition, A shift to the higher angle of the peaks of this perovskite phase is observed upon increasing x (Fig. 5). This corresponds to pseudocubic lattice constant of 3.878, 3.876, 3.874 and 3.873 Å for x = 0, 0.05, 0.10 and 0.15, respectively. The decrease in lattice constant with increasing x is consistent with the substitution of La3+ (higher-valence) by Na1+ (lower-valence) in the perovskite, as discussed below.

XRD patterns of the La1−xNaxCrO3 (0.0 ≤ x ≤ 0.15) samples calcined at 700 °C, (+) Na2CrO4

Evolution of the position of the highest X-ray diffraction peak
The lattice parameters of the perovskites La1−xNaxCrO3 were calculated for each x value from the XRD patterns using Celref program. The values of a, b and c cell parameters versus the degree of substitution x are summarized in Table 1. With the increase of Na content the unit cell volume gradually decreases as seen in the systematic shifts of the diffractions (Fig. 5). The monotonous decrease contrasts with the radius difference between Na1+ (great ionic radius r = 1.39 Å) and La3+ (smaller radius r = 1.36 Å). The volume contraction phenomenon can be explained using the concept of valence states of cations, the substitution of a lower-valence ion on La-sites of LaCrO3 results in a charge-compensating transition of Cr3+ to Cr6+ [10, 42].
As mentioned previously, the larger radii of Na1+ than that of La3+ may cause the increasing of cell volume with x increases. On the other hand, in order to keep equilibrium of charge, the concentration of Cr6+ (0.44 Å) with less radius will increase as x increases, resulting in the contraction of the Cr–O bonds and decrease of the unit cell volume. Figure 6 shows a linear decrease in the unit cell volume with sodium content, indicating that the substitution of sodium in lanthanum site obeys to Vegard’s law. This means clearly that a solid solution in the concentration range (0 ≤ x ≤ 0.1) is obtained at 700 °C.

Lattice volume dependence on sodium content (x) for La1−xNaxCrO3
3.4 Structural and Morphological Characterization
The average crystallite size (D) was evaluated from the broadening of the XRD line width by applying the Scherer’s formula Eq. (1) [43]:
where D is the crystallite size, λ is the wavelength of X-ray, β is the full width at half maximum (β is expressed in radians), and θ is the Bragg’s angle.
The crystallite size obtained from XRD for different compositions is presented in Fig. 7. From this figure, the oxides present an average crystallite size in the range of 28.1–33.1 nm which indicates clearly the nanometric character of these powders. The crystallite size decreases with increasing sodium content. This is probably due to oxygen vacancies induced by the substitution of lanthanum (III) by sodium (I). This result is in good agreement with the decrease of the cell volumes mentioned in Table 1. Similar results are also found for La1−xSrxMnO3 samples synthesized by coprecipitation of precursor acetates [44] and LaCo1−xRuxO3 [45]. It has been shown that the formation of oxygen vacancies leads to a crystal decrease.

Crystallite size of La1−xNaxCrO3 (0.0 ≤ x ≤ 0.15) samples
Table 2 summarizes the basic textural properties of the samples. As noted, the SBET and average pore diametre of the samples monotonically increase with increasing x, while the total pore volume shows a maximum at x = 0.15. This latter suggests that the specific surface area can be determined by the balance between the crystal size and the degree of agglomeration of the nanocrystals in each case. The observed SBET increase can in this sense be basically determined by the crystal size decrease [36].
The grain size of the different oxides calcined at 700 °C, according to PSD measurements, are shown in Fig. 8. The size distribution of the x = 0, 0.05, 0.10 and 0.15 samples shows a maximum at ca. 12.1, 19.4, 14.5 and 16.7 microns, respectively. Size distributions obtained with maximum at tens of microns must result from agglomeration of primary particles. The decrease in the particle size observed upon increasing x contrasts with the evolution observed in SBET values (Table 2). Crystal size estimate from use of the Scherrer equation for the major perovskite phase is of 33.1, 31.7, 29.4 and 28.1 nm for x = 0, 0.05, 0.10 and 0.15, respectively. No direct correlation can be established between these values and those of SBET and pore volume or grain sizes (Table 2; Fig. 8), suggesting that the specific surface area becomes basically determined by morphological properties of the Na doped LaCrO3 nanoheterostructures formed in each case.

Particle size distribution of the samples with x = 0, 0.05, 0.10 and 0.15
The morphologies of the La1−xNaxCrO3 samples examined by SEM are shown in Fig. 9. The micrographs indicate clearly that the shape remains similar which the samples particles are nearly spherical in shape and the powders are partially agglomerated. The average particles sizes of the powders are approximately 80–200 nm. The grain boundaries are compact and there is only very small amount of pores. The formation of agglomerates is probably due to the nature of the solvent used in the preparation of the gels. The average crystallite sizes of the calcined samples determined by X-ray diffraction were significantly smaller than the values determined by SEM. This difference can be attributed to the fact that the values observed by SEM have the size of the secondary particles, which are composed of several or many crystallites by soft reunion.

SEM micrographs of La1−xNaxCrO3. a x = 0, b x = 0.05, c x = 0.1 and d x = 0.15
3.5 Electrochemical Behavior of La1−xNaxCrO3 Electrodes
The electrocatalytic activities of the perovskite oxides were greatly affected by the nature properties of B-site transition metal ions and the concentration of oxygen vacancies [46, 47].
Cyclic voltammograms for the electrodes La1−xNaxCrO3 in 1 M KOH at 25 °C were recorded between −1.5 and 1.5 V at the scan rate 20 mV s−1. Typical voltammograms for the oxygen reduction and evolution reactions of air electrode with different substitution of lanthanum by sodium according to:
are shown in Fig. 10. The voltammograms show the cathodic and anodic current–potential curves of air electrode with different substitutions by sodium. The coated electrode films showed good adherence during polarization. The voltammograms exhibit two redox peaks, an anodic (Epa = 690 mV) and a corresponding cathodic (Epc = 375 mV) peak, prior to the onset of the O2 evolution reaction, revealing a pseudo-capacitance due to the Ni(III)/Ni(II) surface redox couple [48, 49]. At lower current densities (<20 mA cm− 2), the onset potential towards the oxygen evolution reaction is around 0.6 V versus Hg/HgO which is slightly higher than those reported in [50, 51]. All voltammograms show a wide plateau region which indicates that the coated perovskite electrodes present a wide range of electrochemical stability. Both anodic and cathodic scans exhibit a composition dependency of the current density. Indeed, the addition of sodium allowed to raise the current density which is approximately three times higher for La0.85Na0.15CrO3 than LaCrO3 at overpotential of 1 V. It is clear that at high doping level, the sample has a better electrochemical behavior in oxygen reduction and evolution.

Cyclic voltammograms for La1−xNaxCrO3 electrodes in 1 M KOH at 20 mV/s (25 °C). Curves 1–4 correspond to x = 0, x = 0.05, x = 0.10 and x = 0.15, respectively
This is probably due to the structure of Na-containing perovskite. Namely, when lanthanum lattice sites were occupied by sodium ions, negative effective charges, Sr’La, were formed. These negative effective charges must be compensated by the formation of equivalent amount of positive effective charges, such as electron holes (h·) or oxygen vacancies (Vo ·). The presence of these charged defects (h· or Vo ··) in modified electrodes may enhance the ionic conductivity of oxygen ion (O2−) and facilitates the electrocatalytic process. Hence, a better electrocatalytic activity of La0.85Na0.15CrO3 than that of LaCrO3 was expected. A similar result was found in La1−xSrxCoO3 perovskite oxide. In fact, Lal et al. [52] reported that introduction of Sr2+, particularly 0.3 and 0.4 mol in lanthanum lattice sites, increased the apparent electrocatalytic activity of the base oxide about 1.8 times.
4 Conclusion
La1−xNaxCrO3 perovskite-type oxides in the composition range x = 0–0.15 were successfully synthesized by the citrate sol–gel method. TG/DTG and XRD analysis of the solid precursors after treatment in an oven at 100 °C evidences that full decomposition of the organic molecules or intermediate oxides and generation of the lanthanum chromite perovskite phase (according also to FT-IR results) with orthorhombic structure is produced for T ≥ 700 °C. In doped LaCrO3, the solubility limited of Na ions for A-site cation sublattice is less than 15%. The low solubility is probably caused by the ionic radius difference between dopant Na1+ and parent cations. The textural properties of corresponding materials calcined at 700 °C have been explored by SBET measurements and PSD analysis, demonstrating that the agglomeration degree of primary particles increases with increasing amount of sodium. The microstructure and morphology of the compounds show that the samples particles are nearly spherical in shape and the powders are partially agglomerated. The average particle sizes ranging from 80 to 200 nm. The electrochemical behavior carried out by cyclic voltammetry of these samples reveals that the electrode with higher sodium content La0.85Na0.15CrO3 exhibits a higher electroactivity. This indicates that La0.85Na0.15CrO3 oxide is among the investigated series the best electrocatalyst for oxygen evolution and reduction. These results show clearly that there is a close relationship between the activity of the catalyst and the cobalt content.
References
J. Goodenough, W. Graper, F. Holtzberg, D.L. Huber, R.A. Jefever, J.M. Longo, T.R.M. Guire, S. Methfessel, K.H. Hellwege, A.M. Hellwege, in Numerical Data and Functional Relationships in Science and Technology, ed. by Landlot Börnstein (Springer, Berlin, 1970), p. 126
F.S. Galasso, Perovskite and High Tc Superconductors, (Gordon and Breach Science Publishers, New York, 1990)
A. Weber, E. Ivers-Tiffee, J. Power Sources 127, 273 (2004)
H. Hayashi, H. Inaba, M. Matsuyama, N.G. Lan, M. Dokiya, H. Tagawa, Solid State Ion. 122, 1 (1999)
M. Pena, J. Fierro, Chem. Rev 101, 1981 (2001)
H. Taguchi, S.-I. Matsuura, M. Nagao, H. Kido, Phys. B 270, 325 (1999)
F. Boroomand, E. Wessel, H. Bausinger, K. Hippert, Solid State Ion. 129, 251 (2000)
J. Sfeir, P.A. Buffet, P. Mockli, N. Xanthopoulos, R. Vasquez, H.J. Mathieu, J. van-Herle, K.R. Thampi, J. Catal. 202, 229 (2001)
P. Vernoux, E. Djurado, M. Guillodo, J. Am. Ceram. Soc. 10, 2289 (2001)
J.W. Fergus, Solid State Ion. 171, 1 (2004)
D.P. Karim, A.T. Aldred, Phys. Rev. B 20, 2255 (1979)
R. Koc, H.U. Anderson, J. Mater. Sci. 27, 5477 (1979)
T. Nakama, G. Petzow, L.J. Gaukler, Mater. Res. Bull. 14, 649 (1979)
N. Sakai, H. Yokokawa, T. Horita, K. Ymaj, Int. J. Appl. Ceram. Technol. 1, 23 (2004)
A. Zuev, I. Singheiser, K.J. Hilbert, Solid State Ion. 147, 1 (2002)
G.J. Zhang, Y.W. Song, H. Xiong, J.Y. Zheng, Y.Q. Jia, Mater. Chem. Phys. 73, 101 (2002)
K.S. Song, H. Xing-Cui, S.-D. Kim, S.-K. Kang, Catal. Today 47, 155 (1999)
H.P.S. Correa, C.O. Paiva-Santos, L.F. Setz, L.G. Martinez, S.R.H. Mello-Castanhe, M.T.D. Orlando, Powder Diffr. 23, S18–S22 (2008)
W. Kobsiriphat, B.D. Madsen, Y. Wang, M. Shah, L.D. Marks, S.A. Barnet, J. Electrochem. Soc. B 279, 157 (2010)
H. Rahmauni, A. Selmi, K. Khirouni, N. Kallel, J. Alloys Compd. 93, 533 (2012)
X. Liu, W. Su, Z. Lu, L. Pei, W. Liu, L. He, J. Alloys Compd. 21, 305 (2000)
S. Mukherjee, M.R. Gonal, M.K. Patel, M. Roy, A. Patra, A.K. Tyagi, J. Am. Ceram. Soc. 95, 290 (2012)
S.P. Jiang, L. Liu, K.P. Ong, P. Wu, J. Li, J. Pu, J. Power Sources 176, 82 (2008)
Y. Zhang, Q. Zhou, T. He, J. Power Sources 196, 76 (2011)
K.P. Ong, P. Wu, J. Appl. Phys. Lett. 90, 044109 (2007)
V.D. Nithya, R.J. Immanuel, S.T. Senthilkumar, C. Sanjeeviraja, I. Perelshtein, D. Zitoun, R.K. Selvan, Mater. Res. Bull. 47, 1861 (2012)
S. Makhloufi, M. Omari, J. Inorg. Organomet. Polym. 26, 32 (2016)
P.S. Devi, M.S. Rao, Anal. Appl. Pyrolysis 22, 187 (1992)
H. Wang, Y. Zhu, P. Liu, W. Yao, J. Mater. Sci. 38, 1939 (2003)
G. Silva, J. Santos, D. Martinelli, A. Pedrosa, M. de Souza, D. Melo, J. Mater. Sci. Appl. 1, 39 (2010)
A.A. Samat, N.A. Abdullah, M.A.M. Ishak, N. Osman, World Acad. Sci. Eng. Technol. 6, 951 (2012)
K. Rida, A. Benabbas, F. Bouremmad, M.A. Pen, E. Sastre, A. Martinez-Arias, Appl. Catal. A 327, 173 (2007)
X. Ding, Y. Liu, L. Gao, L. Guo, J. Alloys Compd. 458, 346 (2008)
H. Xian, X. Zhang, X. Li, H. Zou, M. Meng, Z. Zou, L. Gou, N. Tsubaki, J. Catal. Today 158, 215 (2010)
Y. Yang, Y. Jiang, Y. Wang, Y. Sun, J. Catal. A. Chem. 270, 56 (2007)
L.G. Tejuca, J.L.G. Fierro, Thermochim. Acta 147, 361 (1989)
A.E. Lavat, E.J. Baran, Vib. Spectrosc. 32(2), 167 (2003)
G.B. Jung, T.J. Huang, M.H. Huang, C.L. Chang, J. Mater. Sci. 36, 5839 (2001)
J.D. Carter, H.U. Anderson, M.G. Shumsky, J. Mater. Sci. 31, 551 (1996)
R.D. Shannon, Acta Crystallogr. Sect. A 32, 751 (1976)
T.-Y. Chen, R.-Y. Pan, K.-Z. Fung, J. Phys. Chem. Solids 69, 540 (2008)
W.Z. Zhu, S.C. Deevi, Mater. Sci. Eng. A 348, 227 (2003)
B.D. Cullity, Elements of X-ray Diffractions, (Addison Wesley Publ, Massachusetts 1978), p. 102
J. Tulloch, S. Donne, J. Power Sources 188, 359 (2009)
N. Mota, M.A. Galvana, R. Navarroa et al., Appl. Catal. B 113, 271 (2012)
K. Kordesch, G. Simader, Fuel Cells and their Applications. (VCH, Weinheim, 1996)
M. Baldauf, W. Preidel, J. Power Sources 84, 161 (1999)
R.N. Singh, J.-F. Koenig, G. Poillerat, P. Chartier, J. Electrochem. Soc. 137, 1408 (1990)
M.A. AbdelRahim, R.M. AbdelHameed, M.W. Khalil, J. Power Sources 134, 160 (2004)
T. Kudo, H. Obayashi, T. Geji, J. Electrochem. Soc. 122, 159 (1975)
M. Shimada, M. Koizumi, J. Appl. Electrochem. 12, 275 (1982)
B. Lal, M.K. Raghunandan, M. Gupta, R.N. Singh, Int. J. Hydrog. Energy 30, 723 (2005)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fodil, H., Omari, M. Synthesis, Structural and Physicochemical Characterization of Nanocrystalline La1−xNaxCrO3 Powders by Sol–Gel Method. J Inorg Organomet Polym 27, 1473–1481 (2017). https://doi.org/10.1007/s10904-017-0605-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-017-0605-x