
Abstract
A modified sol–gel (MSG) method was developed to synthesize perovskite La1−x Sr x MnO3 (x = 0.2, 0.4, 0.6, 0.8) with large specific surface area that allows for control of their particle size from the micrometer level to the nanometer level with novel nanoporous structure. The MSG method utilized carbon black (Vulcan XC-72R) as a pore-forming material during the preparation process. Two important process parameters, the calcination temperature and the pore former material addition, were investigated to control the size of the La1−x Sr x MnO3 particles. The phase evolution of La1−x Sr x MnO3 powders was investigated by thermogravimetric analysis (TG/DSC) and X-ray diffraction pattern. The results showed that the pure La0.6Sr0.4MnO3 phase has been obtained at about 550 °C in air, which is lowered around 150 °C comparing to conventional sol–gel method. In scanning electron microscope studied, it presented novel homogeneous nanoporous morphology with the average particle size from 30 to 100 nm. Based on the Brunauer–Emmett–Teller method analysis, the specific surface area of La1−x Sr x MnO3 was significantly influenced by the calcination temperature, the pore former material addition and the strontium content. The largest specific surface area of La0.2Sr0.8MnO3 reached 114.3 m2/g.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Perovskite-type oxides have been attracting much attention and extensively reviewed their catalytic abilities for oxygen reduction reaction (ORR)/oxygen evolution reaction (OER) in metal-air batteries [1–3] and for combustion of volatile organic compounds in the abatement of exhaust gases [4, 5]. Among them, La1−x Sr x MnO3 (x = 0.2–0.8) materials showed the reasonable catalytic activity for ORR/OER and for combustion of volatile organic compounds [6–8]. As a catalyst, the catalytic activity and stability are prerequisite, strongly relating to its morphology, particle size distribution, and crystallinity, which are determined by the preparation method adopted [9]. Traditionally, the preparation method (e.g., sol–gel [10], citrate complexation [11], solid-state reaction [12], co-precipitation [13]) involve in high-temperature solid-state reactions, leading to the destruction of pore structures and hence to low surface areas (<25 m2/g). That would greatly hinder their practical applications in heterogeneous catalysis. Previously, Kaliaguine et al. [14–16] adopted a reactive grinding method, in which ZnO as additive was ground together with the perovskites-type materials and then leached by NH4NO3, to prepare a series of LaBO3 (B=Co, Mn, and Fe) catalysts with high specific surface area, finding that their catalytic performance for methanol oxidation enhanced with their specific surface area increasing (from 20 to 100 m2/g). However, after the leaching process, a small amount of ZnO still existed and contaminated the perovskite samples, which would reduce the catalytic activities. In order to further increase their catalytic activity, the use of polymeric material as a soft template for the preparation of porous perovskite oxides has also received great interest. Dai et al. [17–19] reported that poly(methyl methacrylate) (PMMA) microspheres are good soft template for the preparation of three dimensionally ordered macroporous (3DOM) perovskite oxides (LaMnO3 and La0.6Sr0.4MnO3), of which macropore sizes and surface areas were 165 nm and 40 m2/g, respectively. And they found that the 3DOM La0.6Sr0.4MnO3 displayed better catalytic performance for methane combustion than that of the bulk La0.6Sr0.4MnO3, due to the larger surface area of 3DOM La0.6Sr0.4MnO3. Nevertheless, it is more complex and difficult for the preparation of soft template with controlled properties in most cases. For the PMMA mentioned above, the preparation and separation of 3-DOM PMMA from the solution is difficult because of the low density of PMMA, thus the separation process (by centrifugal machine) needs high rotational speed and long rotational time. Additionally, the formation of PMMA is sensitive to the environmental conditions and randomly arranged PMMA is usually yielded. To overcome these difficulties, the focus has shifted from increasing its specific surface areas to controlling its morphology. Recently, Jin et al. [20] have successfully synthesized an urchin-like La0.8Sr0.2MnO3 perovskite oxide by co-precipitation method with urea as a precipitator, finding that the urchin-like La0.8Sr0.2MnO3 perovskite oxide exhibits an encouraging catalytic activity for ORR and OER due to its novel morphology. Whereafter, Lu et al. [21] have prepared a hollow spherical La0.8Sr0.2MnO3 (HS-LSM) perovskite oxide using a new carbonate-template route, discovering that the HS-LSM showed high activity and stability for ORR in alkaline solution, outperforming the commercial Pt/C. Although these novel structural perovskite oxides could provide a larger active surface area with reactants relative to the bulk, the particle size and shape of the resulting perovskite oxides is hard to control owing to the complex overall procedures of the preparation methods. Therefore, it is highly desirable to develop an effective facile strategy for the controlled preparation of porous perovskite oxides with high specific surface area.
The aim of this work is to explore a facile modified sol–gel (MSG) method in order to not only reduce the calcination temperature but also increase the surface area of perovskite oxides. A pore-forming material, carbon black Vulcan XC-72R, was introduced into the precursor during preparation by SG method. The MSG method was practical to prepare perovskite La1−x Sr x MnO3 with a high surface area without any additional supporters. The phase evolution of La0.6Sr0.4MnO3 powders has been systematically studied by thermogravimetric analysis (TG/DSC), X-rays diffraction pattern (XRD). Furthermore, the effects of the amount of the pore-forming material and the doped Sr element on the La1−x Sr x MnO3 microstructure and specific surface area were investigated by scanning electron microscope (SEM) and Brunauer–Emmett–Teller (BET), respectively.
2 Experimental
2.1 Preparation of nanoporous La1−x Sr x MnO3 (x = 0.2, 0.4, 0.6, 0.8)
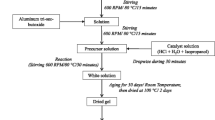
A series of nanoporous La1−x Sr x MnO3 (defined as LSMx, where the x represented different Sr doping content in the samples, x = 0.2, 0.4, 0.6, 0.8) powders were synthesized by a MSG method. Carbon black (Vulcan XC-72R, 30 nm, Cabot) was used as a pore-forming material. La(NO3)3·6H2O (≥99.9 %, Guoyao Chemical Reagent Co. Ltd.), Sr(NO3)2(≥99.0 %, Gouyao Chemical Reagent Co. Ltd.), Mn(NO3)2 (50 wt% aqueous solution, Gouyao Chemical Reagent Co. Ltd.) and citric acid monohydrate (≥99.8 %, Gouyao Chemical Reagent Co. Ltd.) were used as reagents. The key steps are summarized in Fig. 1. Typically, an aqueous solution of citric acid with a 10 % excess mole over the number of ionic equivalents of cations was prepared. The aqueous solution of citric acid was added to various amounts of pore former material, in which the mass ratio of pore former to the theoretical mass of LSMx were 1:1, 2:1 and 3:1, labeled as LSMx-1, LSMx-2, LSMx-3 (x = 0.2, 0.4, 0.6, 0.8). Then they were agitated vigorously for 1 h in room temperature to form homogeneous solution. Subsequently, the aqueous solutions of the stoichiometric metal nitrates were gradually added to the as-prepared solution and they were agitated for another 15 min. After mixture, the resulting solution was stirred vigorously while being heated to 80 °C and kept at this temperature for about 6 h until water nearly completely evaporated and a black dough-like paste was obtained. In order to produce a dry precursor, this paste was dried at 150 °C with a heating rate of 5 °C/min for 2 h in an oven. The precursor was milled to fine powders, which were calcined at various temperatures between 450 and 600 °C with a heating rate of 5 °C/min and maintained for 4 h in air. For comparison, the bulk La1−x Sr x MnO3 samples, marked as B-LSMx (x = 0.2, 0.4, 0.6, 0.8), were prepared by the conventional sol–gel (CSG) method [22, 23]. All synthetic conditions of CSG method were identical to that of MSG method except for the calcinations temperatures (between 600 and 750 °C) and the addition of the pore-forming material.

Flow chart for the preparation of LSMx (x = 0.2, 0.4, 0.6, 0.8) precursors by the MSG method
2.2 Characterization of the as-prepared samples
Phase identifications of the synthesized powders were conducted by a MXPAHF X-ray diffractometer from 20° to 80° with a Cu Kα of 1.54056 Å. Thermogravimetric and differential scanning calorimetry (TG/DSC) was performed with a Netzsch STA 499C in a flow of air (40 cm−3/min) at a heating rate of 10 °C min−1 from room temperature up to 900 °C. Both the morphologies and energy-dispersive X-ray spectroscopy (EDS) of the as-prepared samples were observed by a Hitachi S-4800 SEM equipment. The crystal size of the samples was measured by a Talos F200S transmission electron microscope equipment. The specific surface area was measured by nitrogen adsorption–desorption with the BET method using a NOVA 4200E surface area and pore size analyzer.
3 Results and discussion
Figure 2 shows the TG-DSC curves of the LSM0.4 precursors prepared by the CSG method and the MSG method, which were measured from room temperature to 900 °C in air flow. In the CSG method case, the process could be divided into four stages noted by stage I, II, III and IV in Fig. 2a. There were no peaks in the DSC curve in stage I (around 20–130 °C) corresponding to a mass loss of 2.2 %, which was a consequence of the removal of the residual adsorbed and hydrated water. In stage II (around 130–300 °C), there was an sharp exothermic peak around 150 °C in the DSC curve associating to a mass loss of 54.9 % at this stage, which was due to the loss of nitrate and citric anions by decomposition and oxidation. In stage III (around 300–630 °C), there was an apparent exothermic peak at 328 °C with a mass loss of 23.1 %, which could be assigned to the combustion of metal citrates because the decomposition of metal citrates started at around 320 °C [11]. And the weak exothermic peak at 617 °C is due to formation of the perovskite phase, which is consistent with Ref. [7]. In stage IV (around 630–900 °C), no significant weight loss is observed which implies that almost all the nitrite anion and organic derivatives are removed. In the MSG method case, comparison of the TG and DSC curves showed that the whole process could be divided into five stages noted by stage I, II, III, IV and V in Fig. 2b–d. Compared with the DSC curves of the CSG method, there was an additional strong and sharp exothermal peak between 418 and 580 °C with an obvious mass loss in stage IV when the pore-forming carbon black was introduced during preparation. This was due to the burning combustion of the pore-forming carbon black [24]. In stage V, no significant mass loss was observed, which implied that almost the pore-forming carbon black were removed. As shown in Fig. 2b–d, the onset temperature of no weight loss decreased first and then increased with the increasing amounts of the pore-forming carbon black. For the LSM0.4-1 sample, the crystallization of the perovskite phase is also observed as small endothermic peak around 603 °C in the Fig. 2b. With the pore-forming carbon black content increasing, no noticeable crystallization feature was observed in the DSC profile in Fig. 2c–d. This can probably be attributed to reducing the formation temperature of the perovskite phase in stage IV. And the lowest onset temperature of no weight loss could be reached 510 °C for the LSM0.4-2 sample as shown in Fig. 2c, indicating that the releasing heat in stage IV was beneficial to formation of perovskite phase and decrease the calcination temperature during preparation. While, the onset temperature of no weight loss conversely increased to 580 °C with the further increasing amounts of the pore-forming carbon black, which is due to the formation of the SrCO3 impurity as shown in Fig. 2d.

TG-DSC curves of the LSM0.4 precursors prepared by the CSG method (a) and by the MSG method with the mass ratio of pore former to the theoretical mass of final products (b) 1:1, c 2:1 and d 3:1 for comparison
To obtain information about the formation of crystallographic phase during thermal treatment, XRD analysis was conducted. Figure 3a shows the XRD patterns of the LSM0.4-2 calcined at 450, 500, 550 and 600 °C for 4 h, respectively. For comparison, the XRD patterns of the B-LSM0.4 calcined at 600, 650, 700, 750 °C for 4 h are presented in Fig. 3b. And the standard crystallographic spectrum of perovskite La0.65Sr0.35MnO3 from JCPDS card No. 49-0595 was included. For the LSM0.4-2 samples, a few peaks corresponding to perovskite phase La0.6Sr0.4MnO3 were observed but not well developed at 450 °C, implying that the La0.6Sr0.4MnO3 perovskite phase started to form at 450 °C. Compared with the B-LSM0.4 sample, the peaks of the LSM0.4-2 sample corresponding to perovskite La0.6Sr0.4MnO3 phase were almost observed at 500 °C as shown in Fig. 3a, indicating that perovskite-type oxide La0.6Mn0.4CoO3 phase had already formed. While all diffraction peaks of the B-LSM0.4 sample were indexed as pure La0.6Sr0.4MnO3 phase until the calcination temperature is above 650 °C as shown in Fig. 3b, which was 150 °C higher than that of the LSM0.4-2 sample. This might be ascribed to the introduction of the pore-forming material releasing lots of heat between 350 and 500 °C, which was beneficial to formation of perovskite La0.6Sr0.4MnO3 phase at relatively low calcination temperature. With elevating the calcination temperature, the LSM0.4-2 sample calcined above 550 °C presented the typical pattern of perovskite La0.6Sr0.4MnO3 phase (JCPDS 49-0595) and the corresponding characteristic peaks became sharper as the calcination temperature elevated, demonstrating that better crystalline structure was formed. And no impurities such as manganese oxides, lanthanum oxides and strontium oxides were detected from the XRD patterns.

XRD patterns of LSM0.4 prepared by the MSG method (a) and the CSG method (b) calcined at different temperatures
Figure 4 shows the morphologies of the LSM0.4-2 at different calcination temperatures for 4 h, together with the EDS spectrum of the particles in the selected regions. As shown in Fig. 4a, most of the particles are in round shape with an average particle size of about 30 nm and aggregated to form spongy morphology. The EDS detected amounts of the carbon element (Fig. 4d) confirm that the pore-forming carbon black had not been completely eliminated and still existed in the as-prepared powders when the calcination temperature was 500 °C. With elevating the calcination temperature up to 550 °C, the morphology of the LSM0.4-2 presented a novel nanoporous structure with the pore size of about 100 nm (Fig. 4b). And EDS only detected trace amount of the carbon element as compared Fig. 4e with Fig. 4d, demonstrating that most of the pore-forming carbon black was burnt out, which resulted in homogeneous nanoporous structure in the as-prepared sample. But the particle size of the as-prepared powders increased slightly up to 50 nm than that of the samples calcined at 500 °C (see Fig. 4a). With the calcination temperature further elevating to 600 °C, the LSM0.4-2 powder exhibited particles severely aggregated in platelet shape and the homogeneous nanoporous structure disappeared, owing to excessively high calcination temperature. EDS also did detect trace amount of the carbon element, which might come from the carbon tap during the EDS test. As shown in Fig. 4e, f, the intensity of the carbon element peak almost kept constant, which further suggested that the pore-forming carbon black was completely eliminated at 550 °C. The specific surface area of the LSM0.4-2 samples calcined at 500, 550, 600 °C determined by the BET technique were 71.2, 83.7 and 62.3 m2/g, respectively. It could conclude that excessively high calcination temperature was not conducive to the preparation of the nonporous perovskite oxides with high specific surface area using the MSG method.

SEM images of the LSM0.4-2 calcined at 500 °C (a), 550 °C (b), 600 °C (c) and d–f the EDS spectrum collected onto the square zone of the corresponding images
The influence of the addition amounts of the pore-forming carbon black on the structure and the morphologies of the LSM0.4 samples was investigated by XRD and SEM. Figure 5 presents the XRD patterns of the LSM0.4 prepared by the MSG method with different amounts of the pore-forming carbon black calcined at 550 °C for 4 h. All these three samples displayed the typical pattern of perovskite La0.6Sr0.4MnO3 phase (JCPDS 49-0595), further confirming that the crystallization temperature of the perovskite oxides was greatly reduced when the pore-forming carbon black was introduced. As shown in Fig. 5, the relative intensity of the characteristic peaks for perovskite La0.6Sr0.4MnO3 phase obviously strengthened with the increasing amounts of the pore-forming carbon black. The more pore-forming carbon black was introduced, the more heat it would release, which led to formation better crystalline structure. However, the XRD pattern of the LSM0.4-3 sample appeared impurity peaks at about 25°, 36°, 44° and 50°. This impurity was well indexed as SrCO3, which likely resulted from the reaction of Sr2+ with CO2 (from the combustion of carbon black) during the annealing process. With the increasing amounts of the pore-forming carbon black, the concentration of CO2 gradually increased, promoting the formation of SrCO3. Figure 6 shows the morphologies of the as-synthesized powders with different amounts of the pore-forming carbon black calcined at 550 °C for 4 h. As shown in Fig. 6a, the B-LSM0.4 powders without the pore-forming material, which were obtained by CSG method, exhibited irregular morphology with an average particle size of over 2 μm. When the pore-forming carbon black was introduced, the particle size decreased from the micrometer level to the nanometer level as compared Fig. 6a with Fig. 6b, because the carbon black can hinder the growth of solid particles during heating [25]. Although the particles size of the LSM0.4-1 sample could reached round 100 nm, the particles were severely agglomerated. With the increasing amounts of carbon black, the particle size of LSM0.4-2 sample further reduced to about 50 nm. And the LSM0.4-2 powders exhibited a homogenous novel nonporous structure with the pore size of about 100 nm. The reason is that the carbon black acting as the pore-forming material was burnt out during calcination process and formed the nonporous structure inside the powders. With the further increasing amounts of carbon black, LSM0.4-2 and LSM0.4-3 had the similar particle size as shown in Fig. 6c, d, indicating no influence on morphology with excess addition of carbon black. The specific surface area of B-LSM0.4, LSM0.4-1, LSM0.4-2 and LSM0.4-3 determined by the BET technique were 26.6, 55.6, 83.7 and 112.4 m2/g. The specific surface area of LSM0.4-3 further increased with the amounts of carbon black increasing, manifesting that the more carbon black could produce more nanopores inside the samples. However, the impurity (SrCO3) would be produced when excess amounts of carbon black was added during preparation as shown in the XRD pattern of Fig. 5.

XRD patterns of LSM0.4 prepared by the MSG method with different addition amounts of the pore-forming carbon black

SEM images of LSM0.4 prepared by the MSG method calcined at 550 °C for 4 h b LSM0.4-1, c LSM0.4-2, d LSM0.4-3, and prepared by the CSG method calcined at 700 °C for 4 h: a B-LSM0.4 for comparison
The influence of the ratio of La to Sr on the structure of LSMx powders was investigated by XRD and SEM together with EDS. Figure 7 presents the morphologies and EDS spectrums of LSMx-2 samples with different Sr doping contents prepared by the MSG method calcined at 550 °C for 4 h. Most of the particles are in irregular shape and connected to form a sponge-like morphologies for these four samples. With the increase in Sr content, the sponge-like morphologies remained but the particles size of the samples reduced obviously due to the decrease of unit cell volume with the Sr mole fraction increasing (to be discussed later). For better understanding the actual Sr mole fraction in the samples, the atomic ratio of the transition metals in the samples was determined by EDS, respectively. As shown in Fig. 7, the La/Sr/Mn atomic ratio obtained either from the square zones or from the points were both almost close to the theoretical value, indicating that the Sr content nearly kept constant in the samples after preparation. Figure 8 shows their XRD patterns and the standard crystallographic spectrum of the LaMnO3 parent material based on a cubic unit cell from JCPDS card No. 51-1516. As shown in Fig. 8, the XRD patterns are in good agreement with that reported in literatures for perovskite-type La1−x Sr x MnO3 [7, 26]. Furthermore, there is no the SrCO3 impurity with the increasing Sr content, which was probably due to the relatively low calcination temperature in the MSG case. From the XRD patterns of Fig. 8, the lattice parameters of these four samples could be calculated based on the Pm-3 m space group of the LaMnO3 parent material and the results are summarized in Table 1. Clearly, the unit cell volume decreased with the Sr mole fraction increasing, which was somewhat contrary to the smaller ionic radius of La3+ (117 pm) than that of Sr2+ (132 pm) [28]. The cause of this was that the unit cell size was mainly determined by the manganese ionic radius. With the addition of Sr2+, the manganese oxidation state would change proportionately from Mn3+ to Mn4+, leading to the reduction in the manganese ionic radius from Mn3+ (79 pm) to Mn4+ (67 pm) [6]. With the increase in the ratio of Sr to La, the relative intensity of peaks for LSMx powders gradually reduced implying that the crystallinity of LSMx powders was lowered with increasing Sr content. Moreover, the XRD spectrum of LSMx powders displayed significantly broadening, which might be due to the reduction in the particle size with increasing Sr content. The average crystallite sizes (D) of these four samples were estimated using the Debye–Scherrer equation \((D\left( {hkl} \right) = 0.89\,\lambda /\left( {\beta \,\cos \theta } \right)\), where β is the half-height width of the diffraction peak, θ is the diffraction angle, and λ is the X-ray wavelength) [6, 27]. And their values were about 87, 56, 43, and 32 nm for x = 0.2, 0.4, 0.6, and 0.8, respectively. It was implied that the reduction in the unit cell volume with the increase in the Sr mole fraction led to the crystallite sizes decreased.

SEM images of a LSM0.2-2, b LSM0.4-2, c LSM0.6-2 and d LSM0.8-2 prepared by the MSG method calcined at 550 °C for 4 h and the EDS spectrum collected onto the square zones and points of the corresponding images

XRD patterns of a series of LSMx compounds with x from 0.2 to 0.8 prepared by the MSG method
In order to gain additional supporting evidence, Fig. 9 exhibits the TEM images of LSMx powders for various Sr doping contents. As shown in Fig. 9, all these four samples presented novel nanoporous structure and their average sizes determined by TEM was around 100, 54, 45, and 35 nm for x = 0.2, 0.4, 0.6, and 0.8, respectively, which were in good agreement with that calculated from XRD pattern. This was confirmed that average particle diameter decreased with increasing Sr doping content. The average value lay in the range of 35–100 nm with a fairly narrow size distribution. Furthermore, the specific surface area of the LSMx powders was shown to fall in the range of around 70–110 m2/g and found to be influenced by the Sr doping content. Figure 10 shows a plot of the specific surface area for LSMx powders as a function of the Sr content. As shown in Fig. 10, the specific surface area increased with the Sr doping content, which was consistent with that reported in Ref. [26]. The specific surface area of LSMx were 72.3, 83.7, 96.4 and 114.3 m2/g for x = 0.2, 0.4, 0.6, and 0.8, respectively. The cause of this is that the more nanopores would be produced in the sample with smaller particle size. Obviously, the perovskite-type oxides LSMx, which could be prepared by the MSG method under relatively low calcination temperature (550 °C) condition, demonstrated novel homogeneous nanoporous morphology with the average particle size from 30 to 100 nm. And their specific surface areas surpassed that of LSMx prepared by the CSG method. So, the MSG would be convenient and feasible method to prepare the homogeneous nanoporous oxides with high specific surface area.

TEM images of a LSM0.2-2, b LSM0.4-2, c LSM0.6-2 and d LSM0.8-2 prepared by the MSG method calcined at 550 °C for 4 h

Evolution of the specific surface area as a function of Sr content (x) in LSMx powders
4 Conclusions
The MSG synthesis presented in this paper can be adopted to prepare homogeneous nanoporous structural perovskite-type oxides with control of their particle size from micrometers to the order of tens of nanometers. Based on TG-DSC and XRD results, the pore-forming material were burn out and perovskite phase could be formed at 550 °C. Furthermore, it was observed that the addition of the pore-forming material and the Sr doping content significantly influenced their microstructure and specific surface area. The specific surface area apparently enlarged with the increasing addition of the pore-forming material. However, the SrCO3 impurity was inclined to be produce with excessive pore-forming material. With increasing Sr doping content, the specific surface area of LSMx increased as well. These exciting results provided conclusive evidence that this MSG was a facile and practical method to prepared perovskite-type oxides which possessed homogeneous nanoporous structure and large specific surface area for heterogeneous catalysis.
References
Sapkota P, Kim H (2009) J Ind Eng Chem 15:445–450
Neburchilov V, Wang HJ, Martin JJ, Qu W (2010) J Power Sources 195:1271
Lee YC, Peng PY, Chang WS, Huang CM (2014) J Taiwan Inst Chem Eng 45:2334–2339
Nguyen SV, Szabo V, On DT, Kaliaguine S (2002) Microporous Mesoporous Mater 54:51–61
Deng JG, Zhang L, Dai HX, Au CT (2009) Appl Catal A 352:43–49
Tulloch J, Donne SW (2009) J Power Sources 188:359–366
Wang G, Bao Y, Tian Y, Xia J, Cao D (2010) J Power Sources 195:6463–6467
Liu Y, Dai H, Deng J, Li X, Wang Y, Arandiyan H, Xie S, Yang H, Guo G (2013) J Catal 305:146–153
Zhu J, Li H, Zhong L, Xiao P, Xu X, Yang X, Zhao Z, Li J (2014) ACS Catal 4:2917–2940
Isupova LA, Alikina GM, Tsybulya SV, Boldyreva NN, Kryukova GN, Yakovleva IS, Isupov VP, Sadykov VA (2001) Int J Inorg Mater 3:559–562
Deganello F, Marcì G, Deganello G (2009) J Eur Ceram Soc 29:439–450
Huang L, Zhang F, Wang N, Chen R, Hsu AT (2012) Int J Hydrogen Energy 37:1272–1279
Ma J, Theingi M, Chen Q, Wang W, Liu X, Zhang H (2013) Ceram Int 39:7839–7843
Kaliaguine S, Neste AV, Szabo V, Gallot JE, Bassir M, Muzychuk R (2001) Appl Catal A 209:345–358
Szabo V, Bassir M, Neste AV, Kaliaguine S (2002) Appl Catal B: Environ 37:175–180
Levasseur B, Kaliaguine S (2008) Appl Catal A 343:29–38
Liu Y, Dai H, Du Y, Deng J, Zhang L, Zhao Z (2012) Appl Catal B: Environ 119–120:20–31
Arandiyan H, Dai H, Deng J, Liu Y, Bai B, Wang Y, Li X, Xie S, Li J (2013) J Catal 307:327–339
Liu Y, Dai H, Deng J, Li X, Wang Y, Arandiyan H, Xie S, Yang H, Guo G (2013) J Catal 307:146–153
Jin C, Cao X, Zhang L, Zhang C, Yang R (2013) J Power Sources 241:225–230
Lu F, Sui J, Su J, Jin C, Shen M, Yang R (2014) J Power Sources 271:55–59
Ciambelli P, Cimino S, Rossi SD, Faticanti M, Lisi L, Minelli G, Pettiti I, Porta P, Russo G, Turco M (2000) Appl Catal B: Environ 24:243–253
Wang S, Xue G, Liang J, Yuan Y, Zhang X (2014) Catal Commun 45:39–43
Jakab E, Omastová M (2005) J Anal Appl Pyrolysis 74:204–214
Liu HP, Wang ZX, Li XH, Guo HJ, Peng WJ (2008) J Power Sources 184:469–472
Gaodon M, Laberty-Robert C, Ansart F, Stevens P, Rousset A (2002) Solid State Sci 4:125–133
Gaudon M, Laberty-Robert C, Ansart F, Dessemond L, Stevens P (2004) J Power Sources 133:214–222
Shannon RD, Prewitt CT (1969) Acta Crystallogr B 25:925–946
Acknowledgments
This work was financially supported by the Natural Science Foundation of Fujian Province of China (No. 2014J05064), the Research Foundation of Education Bureau of Fujian Province of China (No. JK2013034) and the Talents Introduction Program of Xiamen University of Technology (No. YKJ12019R).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhuang, S., Liu, Y., Zeng, S. et al. A modified sol–gel method for low-temperature synthesis of homogeneous nanoporous La1−x Sr x MnO3 with large specific surface area. J Sol-Gel Sci Technol 77, 109–118 (2016). https://doi.org/10.1007/s10971-015-3835-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-015-3835-9