
Abstract
A PASE (pot, step, atom, economic) synthetic approach to 5-aryl-6-arylthio-2,2′-bipyridine and 6-arylthio-2,5-diarylpyridine ligands/fluorophores has been reported via SNH in 6-aryl-5H-1,2,4-triazines/aza-Diels-Alder reaction sequence. In this article, the “1,2,4-triazine” methodology was successfully used for the synthesis of C6-thiophenol-substituted (2,2′-bi)pyridines as it is well known that thio-substituted (bi)pyridines and their aza-analogs are of wide practical interest. The photophysical properties of the obtained compounds are studied and compared with those reported earlier for 6-substituted 2,2′-bipyridines. The influence of the nature of substituents in the 6-arylthio(bi)pyridine core on the photophysical properties is discussed. It was observed that the new compounds exhibited promising photophysical properties and could be considered as potential push-pull fluorophores. In addition, they demonstrated greater Stokes shift values compared to the previously described 6-H, 6-arylamino and 6-pentafluoro-2,2′-bipyridines and higher fluorescence quantum yields values compare to pentafluorophenyl-substituted 2,2′-bipyridines. Depending on a nature of (bi)pyridine fluorophore LE (locally excited) and/or ICT (intramolecular charge transfer) state were prevailing in emission spectra.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
2,2′-Bipyridines are perhaps the earliest [1, 2] and most common ligands [3,4,5]. Their fragments are part of bioactive compounds [6] including antitumor ones [7], as well as they are promising fluorophores [8,9,10]. To ensure acceptable photophysical properties, namely, long-wavelength absorption and emission maxima, the presence of an extended π-system in such bipyridines is important.
The main object of this work is arylthio-functionalized derivatives of 5-aryl-2,2′-bipyridines which showed promising photophysical properties [11]. It was demonstrated that the photophysical properties of these fluorophores can be tuned by means of synthetic modification. In particular, C5-aromatic substituents can be modified by means of the introduction of various functionalities, including halogen atoms, phenylethynyl moieties or extra aromatic rings, including annelated ones [6]. The edge pyridine ring of the bipyridine core could also be modified by the introduction of (het)aromatic residues or annelation of aromatic rings [12,13,14]. Modification of the central pyridine ring may also be carried out. In particular, the possibilities of introducing various fragments, such as arenes, phenylethynes [15] as well as the annelation of extra aromatic rings [11] were reported. Additionally, we have demonstrated a possibility of the modification of the central pyridine via so called “1,2,4-triazine methodology” by means of the pre-modification of 1,2,4-triazine precursors by introducing the residues of anilines [16,17,18], aliphatic alcohols [19], and pentafluorobenzene [20] into the C5 position and the following aza-Diels-Alder reaction to afford the C6-substituted 2,2′-bipyridines. In this article, in continuation our research, we wish to report a convenient synthetic approach towards 5-aryl-6-arylthio-2,2′-bipyridine and 6-arylthio-2,5-diarylpyridine fluorophores as well as the studies of photophysical properties of these compounds.
Thio-substituted (bi)pyridines and their aza-analogs are of wide practical interest. For instance thiopyrimidines were reported as GluR5 antagonists [21]. 3-(Perfluoro)arylthio-4,4′-bipyridines were proposed as chalcogen and π-hole donors for the stereoselective HPLC separation purposes [22]. C6-Alkylthio-2,2′-bipyridines were reported as ligands for Сu(II) complexes for light-emitting electrochemical cells [23].
A relatively limited number of synthetic approaches to thio-substituted (bi)pyridines has been reported to date. Namely, the heterocyclization reaction under microwave irradiation can be used [24]. The ipso-substitution of halogen atom in the (bi)pyridine core by the thiophenol residue was also reported, in particular, bromine atom in a pressure vessel conditions [21], the substitution of bromine atom in the presence of sodium hydride [23] or chlorine atom in the presence of alkali [25]. In addition, the Pd-catalyzed cross-coupling between 6-bromobipyridine and thiophenol was reported [26]. Finally, thio(het)arene-substituted-2,2′-bipyridines were obtained via 1,2,4-triazine precursors by the ipso-substitution of chlorine atom(s) by thio(het)arene residues and followed by aza-Diels-Alder reaction sequence [27,28,29].
In terms of the above reported “1,2,4-triazine” methodology, it is worthy to mention a more convenient approach for obtaining multifunctionalized 2,2′-bipyridines via the nucleophilic substitution of hydrogen atom (SNH-reaction) in 1,2,4-triazines and aza-Diels-Alder reaction sequence. Such methodology has previously been successfully used for the synthesis of (bi)pyridines substituted with residues of carboranes [30], acetylenes [31], polynuclear aromatic hydrocarbons [32], coumarins [33], pentafluorobenzene [20], C-H active compounds [34], alcohols [19] and even hydroxyl-group as a way to obtain 2,2′-bipyridin-6-ones [35]. In this article, the “1,2,4-triazine” methodology was successfully used for the synthesis of C6-thiophenol-substituted (2,2′-bi)pyridines.
Results and Discussion
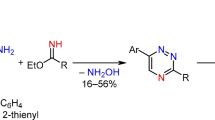
To synthesize the 5-arylthio-3-(2-pyridyl)-1,2,4-triazines the previously reported [36] procedure was initially used via the deoxygenative nucleophilic substitution of hydrogen in 5-H-1,2,4-triazine-4-oxides. However, in case of 3-(2-pyridyl)-1,2,4-triazine-4-oxides 1a-c the following method was followed: namely by a short-time refluxing in acetone in the presence of benzoyl chloride which led to the formation of a mixture of products, probably, due to the reaction of thiophenol both as S- and C-nucleophile. While the same reaction in THF instead of acetone resulted in the formation of the target products 2a-c in up to 78% yields. The same conditions were used for obtaining 5-arylthio-3-(4-fluorophenyl)-1,2,4-triazine 2d, and for obtaining compounds 2e-h the previously described [36] conditions were successfully used.
Further aza-Diels-Alder reaction between the compounds 2 and 2,5-norbornadiene in 1,2-dichlorobenzene in a pressure vessel was carried out as described earlier [37,38,39] which afforded the target 2,2′-bipyridines 3a-d in 75–82% yields (Scheme 1, Fig. 1). Cyclopentene-fused bipyridines 4a-b were prepared via the solvent-free reaction [40] between the corresponding 1,2,4-triazines 2 and 1-morpholinocyclopentene under neat conditions. The purification of the obtained products was performed by means of flash chromatography.

Reagents and conditions: i) 5a-c (R1 = H (a), 3-OMe (b), 4-OMe (c)) THF, CF3COOH, reflux, 30 min, then BzCl, reflux, 30 min; ii) 2,5-norbornadiene, 1,2-dichlorobenzene, 215 °C, 20 h; iv) 1-morpholinocyclopentene, neat, reflux, 20 h.

Chemical structures of fluorophores 3a-d, 4a-b
In a similar way thiophenol-substituted pyridines 3e-h were obtained for studying their photophysical properties with respect to thiophenol-substituted bipyridines 3a-d, 4a-b (Scheme 2).

Reagents and conditions: i) 5a, c (R2 = H (a), OMe (c)) THF, CF3COOH, reflux, 30 min, then BzCl, reflux, 30 min; ii) 5a, d (R2 = H (a), OH (d)), acetone, CF3COOH, reflux, 5 min, then BzCl, reflux, 10 min; iii) 2,5-norbornadiene, 1,2-dichlorobenzene, 215 °C, 20 h.
Photophysical Properties
UV-vis and Photoluminescence (PL) Properties
Spectroscopic measurements for the new fluorophores 3–4 were performed in acetonitrile as an aprotic solvent with an average value of the orientational polarizability (Δf = 0.3). This eliminates the possibility of additional distortion of the results due to protonation. In addition, this reduces the possibility of a bathochromic shift of the absorption and emission maxima (since fluorophores 3–4 as polar compounds) to exhibit a high sensitivity to the polarity of the solvent. Indeed, it is known that upon increasing the solvent polarity the effect of reducing the energy of the excited state is enhanced. That leads radiation at lower energies or, in other words, in a longer wavelength region of a spectra [41]. Absolute quantum yields were measured using the integrating sphere of a Horiba-Fluoromax-4 spectrofluorimeter in CH3CN and acetonitrile.
The non-normalized absorption and emission spectra of ligands 3–4 are shown in Fig. 2, 3 to carry out a comparative analysis of the results.

Absorption spectra (2 × 10−5 M) of the fluorophores 3–4 in acetonitrile

Emission spectra (10−5 M) of the fluorophores 3–4 in acetonitrile
All absorption spectra are similar and contain two broad absorption bands in the near UV region with λmax ~ 280 nm and λmax ~ 325 nm, which corresponds to S0 → S1, S0 → S2 electronic transitions. Characteristic emission spectra of the ligands 3c and 3d are well structured and contain two bands related to both the locally excited (LE) state (λmax = 372 nm (3d), λmax = 364 nm (3c)) and the ICT state (λmax = 443 nm (3d), λmax = 420 nm (3c)). In this case, the Stokes shift values are in the range of 40–50 nm, which leads to the conclusion that the state of fluorophores 3c and 3d predominates in a LE state (the group of the LE state). At the same time, for compounds 4a, 4b, 3a and 3b, the emission spectra are presented as by a continuous unstructured band with a maximum wavelength of 420–440 nm and with an average Stokes shift of about 100 nm. Such characteristics make it possible to attribute them to the group with the predominant intramolecular charge transfer (ICT state group).
As for the fluorescence quantum yields for the compounds 3–4, their values were up to 10.6%, and the maximum value was observed for the compound 4a. Moreover, the introduction of a cyclopentene fragment mostly does not affect the fluorescence quantum yield values. Thus, a comparison of the quantum yields for the related compounds 4a and 3b, as well as 4b and 3c shows no significant difference.
It turned out that the introduction of a condensed cyclopentene fragment into the central pyridine ring in compounds 4 affected the π-conjugation indirectly, i.e., the absorption maxima remained unchanged. But the molar extinction coefficient decreased, while the lifetime of the excited state increased on average by 2.5–3 times. For example, for compound 4a εM = 7500 M−1 cm−1, τ = 7.2 ns, while for compound 3b εM = 9900 M−1 cm−1, τ = 2.7 ns.
LE-State Vs ICT-State for Fluorophores; Solvatochromic Properties
To clarify the nature of intramolecular charge transfer in the excited state and the solvent effect on the fluorescent behavior of chromophores, the study of the general effect of solvents for several fluorophores 3–4 was carried out (Table 2).
In this case, a number of solvents was selected, and the value of the orientation polarizability Δf of the subsequent solvent in the series differed from the previous one by no more than 0.05 units. The largest Stokes shifts of 117 nm for compound 4a were observed in highly polar aprotic solvent DMF and of 114 nm for the compound 4b in a protic solvent methanol. Solvatochromic shift of more than 100 nm usually originates from the redistribution of internal charge transfer in the excited state and a change in the difference between the dipole moments in the ground and excited states of the fluorophore [41]. In order to prove this fact mathematically, the dependence of the Stokes shift values in cm−1 versus the orientation polarizability of solvents were plotted in accordance with the Lippert-Mataga equation.
It turned out that, for fluorophores 3a and 3b, the Lippert-Mataga plots do not obey a linear relationship over the whole range of solvent polarity, and each plot is represented by two independent approximating lines, which indicates the existence of two different excited states (Fig. 4).

Emission spectra (left) of the fluorophore 3a throughout the range of various solvents and Lippert-Mataga plot (right) with two different excitation states (LE-state and ICT-state for 3a)
The transition dipole moment (Δμ) for the compound 3а was calculated from the slope of the Lippert-Mataga plot and amounted to 8.6D in the region of low and medium polar solvents (Δf <0.2) and 13.0D in the region of polar solvents (Δf > 0.2). Approximately similar values were obtained for 3b (8.9D and 12.1D). Thus, high magnitude of the dipole moment (Δμ > 10 D, at R2 > 0.95) for the fluorophores 3a-3b in the region of highly polar solvents indicate a clear predominance of the ICT-state. In this case, the nonlinear relationship between the Stokes shift values and the solvent polarity may indicate an additional intercrossed excited state of these fluorophores. [42]
On the contrary, the solvatochromic shift of the emission maxima for the compounds 3c and 3d was observed only in the non- and low-polarity regions. The high degree of the approximation of both Lippert-Mataga plots (R2 > 0.95) made it possible to determine the difference in dipole moments, which was 4.9D and 7.2D, respectively, which can clearly be attributed to the prevailing LE-state (Fig. 5) and also see the Supplementary Information for compound 3d.

Emission spectra (left) of the fluorophore 3с only in the non- and low-polarity regions (Δf < 0,2) and Lippert-Mataga plot (right) with one excitation state (LE-state)
Despite the regular solvatochromic shift on going from nonpolar cyclohexane to strongly polar DMF, for the compounds 4a and 4b, the permanent dipole moments were 8.3D and 9.7D, respectively, taking into account the low second order approximation reliability (R2 ~ 0.8). At the same time, it was not possible to separate two sections with high approximations in the Lippert-Matag plots. Thus, for the compounds 4a and 4b, the low linear relationship between the Stokes shift values and the solvent polarity, along with low values of transition dipole moment (Δμ < 10D), may indicate a large contribution of both the LE-state in the solvents of low polarity and the ICT-state in the solvents of high polarity due to the hampered π-conjugation caused by the presence of pyridine-fused cyclopentane fragments (Fig. 6).

Emission spectra (left) of the fluorophore 4а in various solvents and Lippert-Mataga plot (right)
We have compared the photophysical properties of new fluorophores with some of their analogs of the 5-aryl-2,2′-bipyridine series having different substituents at position C6. Thus, the introduction of the arylthio group at the position of C6 changes dramatically its photophysical properties of the designed fluorophore in the direction of enhancing “push-pull” properties compared to 6-unsubstituted analogues with or without cyclopentane fragment (Table 1, entry 8) (Table 1, entries 11 and 14). At the same time, the degree of π-conjugation increased significantly, which led to a significant bathochromic shift of the absorption and emission spectra, an increase in the Stokes shift by 1.5–2 times, and also an increase, in some cases, the fluorescence quantum yields. Based on all above, one can conclude that in new chromophores the ICT state is prevailing.
A comparison of the new compounds 3 and 4 with their arylamine-substituted analogs (Table 1, entries 9 and 13) showed that the replacement of the aniline residue with a thiophenol fragment in some cases causes an increase in the Stokes shift, for example, from 89 to 110 nm (entries 1 and 9). At the same time, due to the heavy-atom effect which increases the rate of intersystem crossing [43] the new fluorophores 3 and 4 have lower fluorescence quantum yield values compare to 6-arylamino-substituted 2,2′-bipyridines (2–10% vs up to 48%).
Compare to pentafluorophenyl-substituted 2,2′-bipyridines (Table 1, entries 10, 12, and 15) compounds 3–4 exhibited much higher fluorescence. Thus, the fluorescence quantum yields values for pentafluorophenyl-substituted 2,2′-bipyridines were not exceeding 1%, while for the compounds 3–4 the quantum yield value was observed as high as 10%. The Stokes shift values for pentafluorophenyl-substituted 2,2′-bipyridines were also, as a rule, noticeably lower.
Along with N^N ligands, a photophysical studies of thiophenol-substituted C^N ligands 3e-h were carried out (Scheme 2, Table 3). These compounds demonstrated fluorescence quantum yields values as low as 2.3%, and this is less than the above mentioned N^N ligands 4a-b, 3a-d. 4-Fluorophenyl-substituted pyridines and 5-(4-fluorophenyl) substituted bipyridines are of particular interest as isoelectronic analogs (Fig. 7).

A number of 4-fluorophenyl-substituted pyridines and 5-(4-fluorophenyl) substituted bipyridines and terpy
It is worthy to mention that recently a detailed analysis of the energies of intermolecular interactions and supramolecular architectures of pyridine and fluorobenzene/pentafluorobenzene was carried out [44]. It was shown that the pyridine type nitrogen atom and fluorine atoms as substituents in the aromatic ring induce a molecular arrangement of one type in the solid state which manifested the isotypic transformation principle. It was logical that the replacement of the fluorophenyl group in compound 3e with the 2-pyridyl group in fluorophore 3d did not actually lead to noticeable changes in the photophysical properties of the ligands. On the other hand, the photophysical properties of 6-thiophenol-substituted pyridines were also differ from fluorophenyl-substituted C^N and N^N ligands, including 6-pentafluorophenyl-substituted 2,2′-bipyridine (Table 1, entry 7), whose photophysical properties are very similar to 2,2′:6,2″-terpyridine analogue (terpy).
In conclusion, we have reported a PASE approach to 6-arylthio-2,5-diarylpyridines and 5-aryl-6-arylthio-2,2′-bipyridines, including cyclopentene-fused bipyridines. New compounds exhibited promising photophysical properties and could be considered as potential push-pull fluorophores. In particular, they demonstrated greater Stokes shift values compared to the previously described 6-H, 6-arylamino and 6-pentafluoro-2,2′-bipyridines and higher fluorescence quantum yields values compare to pentafluorophenyl-substituted 2,2′-bipyridines. Depending on a nature of (bi)pyridine fluorophore LE and/or ICT state were prevailing in emission spectra.
Data Availability
The authors declare that the data supporting the findings of this study are available in the article and the supplementary materials.
References
Vasile Scăețeanu G, Chifiriuc M, Bleotu C, Kamerzan C, Măruţescu L, Daniliuc C, Maxim C, Calu L, Olar R, Badea M (2018) Synthesis, structural characterization, antimicrobial activity, and in vitro biocompatibility of new unsaturated carboxylate complexes with 2,2′-bipyridine. Molecules 23:157. https://doi.org/10.3390/molecules23010157
Constable EC, Housecroft CE (2019) The early years of 2,2′-bipyridine—a ligand in its own lifetime. Molecules 24:3951. https://doi.org/10.3390/molecules24213951
Kaes C, Katz A, Hosseini MW (2000) Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units. Chem Rev 100:3553–3590. https://doi.org/10.1021/cr990376z
Hancock RD (2013) The pyridyl group in ligand design for selective metal ion complexation and sensing. Chem Soc Rev 42:1500–1524. https://doi.org/10.1039/c2cs35224a
Fletcher NC (2002) Chiral 2,2′-bipyridines: ligands for asymmetric induction. J Chem Soc, Perkin Trans 1(2002):1831–1842. https://doi.org/10.1039/B204272J
Izuta S, Kosaka S, Kawai M, Miyano R, Matsuo H, Matsumoto A, Nonaka K, Takahashi Y, Omura S, Nakashima T (2018) Dipyrimicin a and B, microbial compounds isolated from Amycolatopsis sp. K16-0194. J Antibiot (Tokyo) 71:535–537. https://doi.org/10.1038/s41429-018-0028-0
Bertrand B, Casini A (2014) A golden future in medicinal inorganic chemistry: the promise of anticancer gold organometallic compounds. Dalt Trans 43:4209–4219. https://doi.org/10.1039/C3DT52524D
Loren JC, Siegel JS (2001) Synthesis and fluorescence properties of manisyl-substituted terpyridine, bipyridine, and phenanthroline. Angew Chemie Int Ed 40:754–757. https://doi.org/10.1002/1521-3773(20010216)40:4<754::AID-ANIE7540>3.0.CO;2-T
Karnahl M, Krieck S, Görls H, Tschierlei S, Schmitt M, Popp J, Chartrand D, Hanan GS, Groarke R, Vos JG, Rau S (2009) Synthesis and photophysical properties of 3,8-disubstituted 1,10-phenanthrolines and their ruthenium (II) complexes. Eur J Inorg Chem 2009:4962–4971. https://doi.org/10.1002/ejic.200900310
Krinochkin AP, Kopchuk DS, Chepchugov NV, Kim GA, Kovalev IS, Rahman M, Zyryanov GV, Majee A, Rusinov VL, Chupakhin ON (2017) An efficient synthetic approach towards new 5,5′-diaryl-2,2′-bipyridine-based fluorophores. Chinese Chem Lett 28:1099–1103. https://doi.org/10.1016/j.cclet.2016.12.043
Kozhevnikov VN, Shabunina OV, Kopchuk DS, Ustinova MM, König B, Kozhevnikov DN (2008) Facile synthesis of 6-aryl-3-pyridyl-1,2,4-triazines as a key step toward highly fluorescent 5-substituted bipyridines and their Zn (II) and Ru (II) complexes. Tetrahedron 64:8963–8973. https://doi.org/10.1016/j.tet.2008.06.040
Kopchuk DS, Khasanov AF, Kim GA, Nosova EV, Zyryanov GV, Kovalev IS, Rusinov VL, Chupakhin ON (2015) Functionalized 2-(5-arylpyridin-2-yl)quinolines: synthesis and photophysical properties. Russ Chem Bull 64:872–877. https://doi.org/10.1007/s11172-015-0947-x
Starnovskaya ES, Kopchuk DS, Khasanov AF, Tanya OS, Santra S, Giri K, Rahman M, Kovalev IS, Zyryanov GV, Majee A, Charushin VN (2019) Synthesis and photophysics of new unsymmetrically substituted 5,5′-diaryl-2,2′-bypiridine-based “push-pull” fluorophores. Dyes Pigments 162:324–330. https://doi.org/10.1016/j.dyepig.2018.10.040
Kopchuk DS, Chepchugov NV, Starnovskaya ES, Khasanov AF, Krinochkin AP, Santra S, Zyryanov GV, Das P, Majee A, Rusinov VL, Charushin VN (2019) Synthesis and optical properties of new 2-(5-arylpyridine-2-yl)-6-(het)arylquinoline-based “push-pull” fluorophores. Dyes Pigments 167:151–156. https://doi.org/10.1016/j.dyepig.2019.04.029
Shabunina OV, Kapustina DY, Krinochkin AP, Kim GA, Kopchuk DS, Zyryanov GV, Li F, Chupakhin ON (2017) π-Extended fluorophores based on 5-aryl-2,2′-bipyridines: synthesis and photophysical studies. Mendeleev Commun 27:602–604. https://doi.org/10.1016/j.mencom.2017.11.021
Kopchuk DS, Chepchugov NV, Kovalev IS, Santra S, Rahman M, Giri K, Zyryanov GV, Majee A, Charushin VN, Chupakhin ON (2017) Solvent-free synthesis of 5-(aryl/alkyl)amino-1,2,4-triazines and α-arylamino-2,2′-bipyridines with greener prospects. RSC Adv 7:9610–9619. https://doi.org/10.1039/c6ra26305d
Kopchuk DS, Krinochkin AP, Starnovskaya ES, Shtaitz YK, Khasanov AF, Taniya OS, Santra S, Zyryanov GV, Majee A, Rusinov VL, Chupakhin ON (2018) 6-Arylamino-2,2′-bipyridine “push-pull” Fluorophores: solvent-free synthesis and Photophysical studies. ChemistrySelect 3:4141–4146. https://doi.org/10.1002/slct.201800220
Starnovskaya ES, Shtaitz YK, Krinochkin AP, Khasanov AF, Kopchuk DS, Zyryanov GV, Rusinov VL, Chupakhin ON (2019) The synthesis of 6-phenoxyphenylamino-2,2′-bipyridines as new fluorophores. AIP Conf Proc 2063:040056. https://doi.org/10.1063/1.5087388
Savchuk MI, Khasanov AF, Kopchuk DS, Krinochkin AP, Nikonov IL, Starnovskaya ES, Shtaitz YK, Kovalev IS, Zyryanov GV, Chupakhin ON (2019) New push-pull fluorophores on the basis of 6-alkoxy-2,2′-bipyridines: rational synthetic approach and photophysical properties. Chem Heterocycl Compd 55:554–559. https://doi.org/10.1007/s10593-019-02495-5
Moseev TD, Varaksin MV, Gorlov DA, Nikiforov EA, Kopchuk DS, Starnovskaya ES, Khasanov AF, Zyryanov GV, Charushin VN, Chupakhin ON (2019) Direct C H/C Li coupling of 1,2,4-triazines with C6F5Li followed by aza-Diels-Alder reaction as a pot, atom, and step economy (PASE) approach towards novel fluorinated 2,2′-bipyridine fluorophores. J Fluor Chem 224:89–99. https://doi.org/10.1016/j.jfluchem.2019.05.008
Hammerland LG, Johansson M, Malmström J, Mattsson JP, Minidis ABE, Nilsson K, Peterson A, Wensbo D, Wållberg A, Österlund K (2006) Structure-activity relationship of thiopyrimidines as mGluR5 antagonists. Bioorg Med Chem Lett 16:2467–2469. https://doi.org/10.1016/j.bmcl.2006.01.100
Peluso P, Gatti C, Dessì A, Dallocchio R, Weiss R, Aubert E, Pale P, Cossu S, Mamane V (2018) Enantioseparation of fluorinated 3-arylthio-4,4′-bipyridines: insights into chalcogen and π-hole bonds in high-performance liquid chromatography. J Chromatogr A 1567:119–129. https://doi.org/10.1016/j.chroma.2018.06.060
Alkan-Zambada M, Keller S, Martínez-Sarti L, Prescimone A, Junquera-Hernández JM, Constable EC, Bolink HJ, Sessolo M, Ortí E, Housecroft CE (2018) [cu(P^P)(N^N)][PF6] compounds with bis (phosphane) and 6-alkoxy, 6-alkylthio, 6-phenyloxy and 6-phenylthio-substituted 2,2′-bipyridine ligands for light-emitting electrochemical cells. J Mater Chem C 6:8460–8471. https://doi.org/10.1039/c8tc02882f
Wang XH, Cao XD, Tu SJ, Zhang XH, Hao WJ, Yan S, Wu SS, Han ZG, Shi F (2009) An efficient and direct synthesis of 2-thiopyridines via microwave-assisted three-component reaction. J Heterocycl Chem 46:886–889. https://doi.org/10.1002/jhet.161
Buhleier E, Vögtle F (1977) Ligandstruktur und komplexierung, IX Überbrückte phenanthroline und bipyridine Justus Liebigs Ann Chem 1977:1080–1086. https://doi.org/10.1002/jlac.197719770703
Carroll J, Woolard HG, Mroz R, Nason CA, Huo S (2016) Regiospecific acylation of cycloplatinated complexes: scope, limitations, and mechanistic implications. Organometallics 35:1313–1322. https://doi.org/10.1021/acs.organomet.6b00174
Ławecka J, Olender E, Karczmarzyk Z, Wysocki W, Branowska D, Urbańczyk-Lipkowska Z, Kalicki P (2014) A convenient synthesis of 5,5′-bi-1,2,4-triazines via direct S-arylation and its application in the synthesis of 2,2′-bipyridines. Heterocycl Commun 20:5–9. https://doi.org/10.1515/hc-2013-0214
Branowska D (2005) A direct route to 6,6′-disubstituted-2,2′-bipyridines by double Diels-Alder/retro Diels-Alder reaction of 5,5′-bi-1,2,4-triazines. Molecules 10:274–278. https://doi.org/10.3390/10010274
Branowska D, Rykowski A, Wysocki W (2005) A facile S-transalkylation of 2,2′-bipyridine alkyl sulfides - a new tool for the synthesis of annulated biheterocycles. Tetrahedron Lett 46:6223–6226. https://doi.org/10.1016/j.tetlet.2005.07.062
Prokhorov AM, Kozhevnikov DN, Rusinov VL, Chupakhin ON, Glukhov IV, Antipin MY, Kazheva ON, Chekhlov AN, Dyachenko OA (2006) Carborane-functionalized polyaza aromatic ligands: synthesis, crystal structure and a copper (II) complex. Organometallics 25:2972–2977. https://doi.org/10.1021/om051058v
Kozhevnikov DN, Kozhevnikov VN, Prokhorov AM, Ustinova MM, Rusinov VL, Chupakhin ON, Aleksandrov GG, König B (2006) Consecutive nucleophilic substitution and aza Diels-Alder reaction - an efficient strategy to functionalized 2,2′-bipyridines. Tetrahedron Lett 47:869–872. https://doi.org/10.1016/j.tetlet.2005.12.006
Kovalev IS, Kopchuk DS, Khasanov AF, Zyryanov GV, Rusinov VL, Chupakhin ON (2014) The synthesis of polyarene-modified 5-phenyl-2,2′-bipyridines via the methodology and aza-Diels-Alder reaction. Mendeleev Commun 24:117–118. https://doi.org/10.1016/j.mencom.2014.03.018
Fatykhov RF, Savchuk MI, Starnovskaya ES, Bobkina MV, Kopchuk DS, Nosova EV, Zyryanov GV, Khalymbadzha IA, Chupakhin ON, Charushin VN, Kartsev VG (2019) Nucleophilic substitution of hydrogen–the Boger reaction sequence as an approach towards 8-(pyridin-2-yl)coumarins. Mendeleev Commun 29:299–300. https://doi.org/10.1016/j.mencom.2019.05.019
Shtaitz YK, Savchuk MI, Starnovskaya ES, Krinochkin AP, Kopchuk DS, Santra S, Zyryanov GV, Rusinov VL, Chupakhin ON (2019) Synthesis of 2-phenyl-2-(5-phenyl-2,2′-bipyridin-6-yl)-acetonitrile by “1,2,4-triazine” method with using autoclave. AIP Conf Proc 2063:040050. https://doi.org/10.1063/1.5087382
Savchuk MI, Shtaitz YK, Kopchuk DS, Zyryanov GV, Eltsov OS, Pospelova T, Rusinov VL, Chupakhin ON (2019) Efficient one-step synthesis of 3-aryl-2-pyridones from 6-aryl-1,2,4-triazin-5-ones. Chem Heterocycl Compd 55:985–988. https://doi.org/10.1007/s10593-019-02566-7
Kozhevnikov DN, Kovalev IS, Rusinov VL, Chupakhin ON (2001) SNH reactions of 1,2,4-triazine N-oxides, pyrazine N-oxides, and pterin N-oxides with arenethiols. Russ Chem Bull 50:1068–1071. https://doi.org/10.1023/A:1011385706308
Savchuk MI, Starnovskaya ES, Shtaitz YK, Kopchuk DS, Nosova EV, Zyryanov GV, Rusinov VL, Chupakhin ON (2018) Synthesis of 5-phenyl-2,2′-bipyridines 6-substituted with donor groups by aza-Diels–Alder reactions of 5-R-1,2,4-triazines under high pressure conditions. Russ J Gen Chem 88:2213–2215. https://doi.org/10.1134/S1070363218100316
Kumar NSS, Shafikov MZ, Whitwood AC, Donnio B, Karadakov PB, Kozhevnikov VN, Bruce DW (2016) Mesomorphism and photophysics of some metallomesogens based on hexasubstituted 2,2′:6′, 2′′-terpyridines. Chem Eur J 22:8215–8233. https://doi.org/10.1002/chem.201505072
Krayushkin MM, Sedishev IP, Yarovenko VN, Zavarzin IV, Kotovskaya SK, Kozhevnikov DN, Charushin VN (2008) Synthesis of pyridines from 1,2,4-triazines under high pressure. Russ J Org Chem 44:407–411. https://doi.org/10.1134/s1070428008030160
Kozhevnikov VN, Ustinova MM, Slepukhin PA, Santoro A, Bruce DW, Kozhevnikov DN (2008) From 1,2,4-triazines towards substituted pyridines and their cyclometallated Pt complexes. Tetrahedron Lett 49:4096–4098. https://doi.org/10.1016/j.tetlet.2008.04.138
Lakowicz JR (2006) Principles of fluorescence spectroscopy. Springer, Springer US, Boston, MA https://doi.org/10.1007/978-0-387-46312-4
Song H, Wang K, Kuang Z, Sheng Zhao Y, Guo Q, Xia A (2019) Solvent modulated excited state processes of push–pull molecule with hybridized local excitation and intramolecular charge transfer character. Phys Chem Chem Phys 21:3894–3902. https://doi.org/10.1039/c8cp06459h
Cui G, Fang WH (2013) State-specific heavy-atom effect on intersystem crossing processes in 2-thiothymine: a potential photodynamic therapy photosensitizer. J Chem Phys 138:044315. https://doi.org/10.1063/1.4776261
Shishkin OV, Merz K, Vasylyeva V, Zubatyuk RI (2018) Isotypic transformation principle in molecular crystals. Analysis of supramolecular architecture of fluorinated benzenes and pyridines. Cryst Growth Des 18:4445–4448. https://doi.org/10.1021/acs.cgd.8b00432
Code Availability
ChemDraw, Origin.
Funding
This work was supported by the Grants Council of the President of the Russian Federation (no. NSh-2700.2020.3), by the Russian Science Foundation (Grant # 19–73-10144) and State Contract # (0398–2019-0001 АААА-А19–119011790132-7).
Author information
Authors and Affiliations
Contributions
Maria I. Savchuk: Data curation, Investigation, Methodology. Dmitry S. Kopchuk: Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Supervision, Writing original draft. Olga S. Taniya: Formal analysis, Writing original draft, review, and editing. Igor L. Nikonov: Data curation, Formal analysis, Investigation, Methodology. Ilya N. Egorov: Investigation, Methodology. Sougata Santra: Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Writing original draft review and editing. Grigory V. Zyryanov: Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Writing original draft, review and editing. Oleg N. Chupakhin: Conceptualization, Project administration, Supervision. Valery N. Charushin: Conceptualization, Project administration, Supervision.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest/Competing Interests
The authors declare that they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOC 5280 kb)
Rights and permissions
About this article

Cite this article
Savchuk, M.I., Kopchuk, D.S., Taniya, O.S. et al. 5-Aryl-6-arylthio-2,2′-bipyridine and 6-Arylthio-2,5-diarylpyridine Fluorophores: Pot, Atom, Step Economic (PASE) Synthesis and Photophysical Studies. J Fluoresc 31, 1099–1111 (2021). https://doi.org/10.1007/s10895-021-02714-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-021-02714-3