

The method for the preparation of 3,6-disubstituted 1,2,4-triazines based on condensation reaction between easily available α-imino esters and isonitrosoacetophenone hydrazones was reported. The significant differences in reaction conditions, ratio of products, and yields between the developed method and the earlier reported approach were demonstrated. The corresponding 2,5-disubstituted pyridines were synthesized from the prepared 1,2,4-triazines, and their photophysical properties were studied. Studies of the photo-physical properties revealed low and moderate luminescence quantum yields, and negligible solvatochromic behavior in case of 4-methoxyphenylpyridine derivative due to the role of donating methoxy group, however, with a low linearity of a Lippert–Mataga plot. Nevertheless, 2,5-disubstituted pyridines are of interest due to simple protocols of synthesis, moderate photophysical properties, and potential applicability in different scientific and industrial areas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Pyridine is one of the most common, widespread, and applicable N-heterocycle that could be found in natural products1 and pharmaceuticals.2 It also can be applied as a ligand for different metal cations. Particularly, 2,5-disubstituted pyridines can find their application as ligands in platinum3 and iridium4 complexes, including the ones for the production of white OLEDs,5 pyridine-boron complexes,6,7 herbicides,8,9 and DNA-binding agents.10 Thus, 2,5-disubstituted pyridines are of significant practical interest.
One of the methods for the synthesis of substituted pyridines is the transformation of the 1,2,4-triazine ring into a pyridine one in an aza-Diels–Alder reaction with various dienophiles.11,12,13 Previously, 2,5-diaryl-substituted pyridines were synthesized using this method.14,15 The starting substrates in this case should be 3,6-disubstituted 1,2,4-triazines; the approaches to their synthesis were considered by us earlier.16 Among these methods, our attention was drawn to condensations using readily available isonitrosoacetophenone hydrazones. The second substrate for the condensation reaction can be aldehyde17,18 or ortho ester.19 In addition, there is a known protocol that describes the use of α-imino esters derived from the corresponding nitriles. This method is attractive because nitriles are often more accessible than the corresponding aldehydes and ortho esters. However, despite the attractiveness of this method, it has been developed to an insignificant extent and is only presented in a few works.14,20,21,22,23 In some cases, this method leads to the production of 1,2,4-triazine 4-oxides. As for 1,2,4-triazines, this method has been used to prepare compounds containing various pyridyl residues or its aza/fused analogs, as well as di- or trichloromethyl group at the C-3 position. In this work, we have proposed a convenient approach for the preparation of 1,2,4-triazines functionalized at the C-3 position with different (hetero)aromatic or aliphatic substituents.
α-Imino esters 2a–e were obtained as hydrochlorides from the corresponding nitriles (Scheme 1).24 During the heterocyclization reaction, α-imino esters were released in free form in situ as a result of interaction with sodium methoxide. The further reaction was carried out according to the previously reported procedure.14 As a result, two products were found in the reaction mixture in almost all cases: the corresponding 1,2,4-triazines 3 and their N-oxides 4. These two products could be separated using column chromatography (Scheme 1, paths a and b, intermediates A–D are shown according to the previously published mechanism).19 1,2,4-Triazines 3b–j were obtained with the yields of 16–56%, while 1,2,4-triazine 4-oxides 4a–j were obtained in 6–62% yields. When α-imino esters 2b–d with an aromatic substituent were used, triazines 3b–e,g–i predominated in the products mixture, while the corresponding N-oxides 4b–e,g–i were only byproducts (Table 1). The situation changed when the aromatic fragment was replaced by an electron-donating 2-thienyl. In this case, the amount of triazine 4-oxides 4f,j increased significantly (yield up to 43%), and they actually became the main reaction products. When methyl- containing α-imino ester 2a was used, N-oxide 4a was generally the only product.

The synthesis of 1,2,4-triazines 3b–j, N-oxides 4a–j, and 2,5-disubstituted pyridines 5b–j and 6c,f
Previously, a similar pattern was studied that describes the influence of the electron-withdrawing ability of a substituent in an α-imino ester on the course of the reaction in the series of mono-, di-, and trichloromethyl-substituted esters.21 Obviously, if an α-imino ester contains the methyl group, which is significantly more electron-donating than chloromethyl group, the exclusive formation of the triazine 4-oxide 4 occurs.
It should be noted that similar heterocyclization with aromatic α-imino esters was previously reported in a single publication,23 where the corresponding 1,2,4-triazine 4-oxides were the only products obtained with yields of 16– 54%. In this case, the authors used α-imino esters derived from substituted benzonitriles that contained both an electron-donating methoxy group and an electron-withdrawing nitro group in the para position. Perhaps, the reason for the exclusive formation of N-oxides was different reaction conditions, specifically without preliminary removal of the hydrochloride, as well as with a much longer reaction time. Thus, the authors dissolved two starting compounds in absolute methanol and kept them for 12 h to 3 days at room temperature or at –20°C. The resulting precipitate was purified by recrystallization (in some cases repeated) from various solvents such as CHCl3, DMSO, or a mixture of DMF–H2O, 8:1. Authors also tested different molar ratios of hydrazone and α-imino ester (from 1:1 to 1:2.5).23
We attempted to reproduce this technique in the reaction of isonitroso-4(3)-fluoroacetophenone hydrazones 1c,d with α-imino ester hydrochloride 2c. However, after keeping the solution of the substrates in methanol at room temperature for 1 day or 3 days, we did not detect the formation of products 3d,h or 4d,h. The analysis of the reaction mixture by NMR revealed only mixtures of unidentifiable products, likely nonaromatized intermediates A–D (Scheme 1). Their brief refluxing in AcOH resulted in a mixture of the corresponding 1,2,4-triazines 3d,h and their N-oxides 4d,h in ratios of 30/70 and 50/50, respectively. The yields of N-oxides 4d,h were 50 and 31%, respectively. In this case, prolonged exposure of the reagents increased the proportion of triazine 4-oxides 4d,h in the product mixture. However, triazines 3d,h, were still formed. In other words, the formation of a mixture of 1,2,4-triazine and 1,2,4-triazine 4-oxide occurs in all cases, even including the one described in the publication.23 Thus, it is likely that the authors of article23 misinterpreted the results of this reaction. At the same time, the following facts attract attention: in their procedure there is no stage of aromatization of products, and they also use recrystallization (often multiple) to isolate products. That is, perhaps, at the recrystallization stage, aromatization of the triazine ring occurs as a result of increasing the temperature, as well as separation of the second product (i.e., 1,2,4-triazine, the nature of which was not analyzed by the authors). Repeating this procedure multiple times is probably required to completely separate the triazine.
The structure of 1,2,4-triazines 3b–j and their N-oxides 4a–j was confirmed based on the 1H, 19F, and 13C NMR data, mass spectrometry, and elemental analysis. It should be noted that the 1H NMR spectra of products 3b–j and 4a–j are quite similar, and the mass spectroscopy and elemental analysis data are especially necessary to establish their structure accurately. In addition, in a number of cases, the structure of triazine 4-oxides (in particular, 4c,d,f) were confirmed by an alternative method based on the condensation of isonitrosoacetophenone hydrazones 1 with aldehydes followed by oxidative aromatization (Scheme 1, method III).17 1,2,4-Triazine 3b was described previously25 (the synthesis was performed by an alternative method), and the spectral data obtained by us coincided with the published ones.
Additionally, the structure of products 3b and 4e was confirmed by X-ray diffraction analysis. According to the crystallography data, two independent molecules of compound 3b are crystallized in the centrosymmetric space group of the monoclinic system (Fig. 1). The molecules are distinguished by torsion angles between phenyl substituent and triazine ring and demonstrate nonsignificant deviations in the bond distance and interatomic angles. In general, the geometry of the molecules is near to standard. Any significantly shortened intermolecular contacts in the molecules were not observed.

Molecular structure of compound 3b in the thermal ellipsoids of the 50% probability level.
Compound 4e crystallized in the centrosymmetric space group of the monoclinic system. The bond distances and angles in the molecule are near to expectation (Fig. 2). The molecule is nonplanar, the torsion angles N(4)–C(3)–C(13)–C(18) 14.7(5)°, N(1)–C(6)–C(7)–C(8) 14.1(5)°. The O atom of the NO group forms the weak H-bonds with C(5)–H(5) and C(12)–H(12) atoms of the nearest molecule (1.5 – x, y – 0.5, 0.5 – z) (Fig. 3). The π–π interactions are observed in the crystal between the triazine ring and the 4-MeOC6H4 moiety of the nearest molecule (x, y – 1, z) (Fig. 4).

Molecular structure of compound 4e in the thermal ellipsoids of the 50% probability level.

The shortened contacts in the single crystal of molecule 4e (in Å).

The π–π interactions in a single crystal of molecule 4e (in Å).
Further aza-Diels–Alder reaction of 1,2,4-triazines 3b–j with 2,5-norbornadiene where carried out in accordance with the previously described procedure,12 this allowed us to obtain the target 2,5-disubstituted pyridines 5b–j in yields up to 68% (Scheme 1). In addition to 2,5-norbornadiene, we also used another well-known dienophile – 1-morpholinocyclopentene, that provided respective cyclopentane-annulated pyridines 6c,f under solvent-free conditions26 with the yield up to 48%. The structures of the obtained compounds 5b–j and 6c,f were confirmed by 1H, 19F, and 13C NMR spectroscopy, mass spectrometry, and elemental analysis. In particular, in the 1H NMR spectra of compounds 5b–j one can note the appearance of signals from the protons of the ABX system of the new pyridine ring in the form of two doublets and a doublet of doublets with the corresponding coupling constants, as well as a high-field shift of the signals of a number of protons in the substituents of the former triazine ring. For annulated pyridines 6c,f, one can note the appearance of signals of the cyclopentane fragment in the resonance region of aliphatic protons and carbons in the 1H and 13C NMR spectra, respectively. In the case of compound 5d, one can note the coincidence of the previously published27 spectral data with those obtained by us. Also, the structure of compound 5g was confirmed by X-ray diffraction analysis. According to X-ray diffraction data, compound 5g is crystallized in the centrosymmetric space group of the orthorhombic system. The structure is nonplanar (Fig. 5), the bond distances and angles in the molecules are near to expectations. In the crystal, the shortened T-like contacts C–H···CAr are observed (Fig. 6).

Molecular structure of compound 5g in the thermal ellipsoids of the 50% probability level. The figure shows torsion angles in degrees.

The shortened contacts in the single crystal of molecule 5g (in Å).
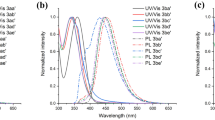
The photophysical properties for the synthesized compounds were studied (Table 2, Fig. 7). In acetonitrile solution, the absorption spectrum maxima are in the range of 287–321 nm, the fluorescence maxima are in the range of 347–375 nm, the Stokes shift is 49–72 nm, and the luminescence quantum yields vary in the range from less than 0.1 to 52.3% depending on the type and positions of substituents. For example, the highest Stokes shift was found for compound 6c, containing a single methoxy group in the phenyl substituent in cyclopentene-annulated pyridine, and the highest quantum yield of luminescence was found in case of compound 5e, functionalized with 4-methoxyphenyl and 4-fluorophenyl moieties. Pyridines 6c,f were chosen as model compounds for solvatochromism studies (Fig. S1 and Table S2, Fig. S2 and Table S3, respectively, Supplementary information file). It was found that in the case of pyridine 6c the difference in the emission maxima between the least and most polar solvents was 25 nm (due to a donating nature of the methoxy group28), while in the case of pyridine 6f – 4 nm. Both values are low, as evidenced by the low linearity of the Lippert–Mataga plot (r2 < 0.14), as well as in comparison with 2,2'-bipyridine analogs, which have been previously described by our scientific group in many works.29,30 However, pyridines 5b–j and 6c,f are compare favorably in terms of ease of synthesis, good product yields, and moderate/high quantum yields of luminescence (depending on the nature and positions of the substituents).

a) UV/Vis absorption spectra and b) normalized emission spectra of compounds 5b–j and 6c,f in MeCN at room temperature at concentration 10–5 M.
In summary, the method for preparation of 3,6-disubstituted 1,2,4-triazines and their N-oxides based on condensation reaction between easily available α-imino esters and isonitrosoacetophenone hydrozones has been reported. A comparison between the developed method and the one reported earlier in literature was conducted. The ratios of the yields of the synthesized 1,2,4-triazines and their corresponding 1,2,4-triazine 4-oxides were determined, and the course of reaction of the developed method and the one reported in literature was discussed. 2,5-Disubstituted pyridines and their cyclopentene-annulated analogs were synthesized from the obtained 1,2,4-triazines in aza-Diels–Alder reaction in good yields. Despite unremarkable photo-physical properties (except high quantum yield of luminescence for one substituted pyridine) and low solvatochromic behavior, some 2,5-disubstituted pyridines could find a promising application due to simple synthetic protocols of their preparation.
Experimental
1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance-400 spectrometer (400, 101, and 376 MHz, respectively), 298K, digital resolution ± 0.01 ppm, using as internal standards TMS (for 1H, 13C NMR spectra) and CFCl3 (for 19F NMR spectra) and C6F6 as internal standard for 19F spectrum of compound 5i. UV-Vis spectra were recorded on a Shimadzu UV1800 spectrophotometer. Luminescence spectra were recorded on a Horiba-Fluoromax-4 spectrofluorometer equipped with integrated sphere. Mass spectrometric studies were performed on an Agilent 6545 Q-TOF LC/MS (Agilent Technologies, USA) quadrupole time-of-flight mass spectrometer with an electrospray ionization source in the positive (negative) ion mode. An Agilent 1290 Infinity II chromatographic system was used to inject the sample. Elemental analysis was performed on a PE 2400 II CHN-analyzer (PerkinElmer).
All reagents were purchased from commercial sources and used without further purification. Silica gel 60 (Kiesel-gel 60, 230–400 mesh) was used for the column chromatography.
The starting hydrazones 1a–c18 and α-imino ester hydrochlorides24 were synthesized as described in literature. The starting hydrazine 1d was synthesized according to method described in the literature for similar compounds.18 1,2,4-Triazine 4-oxides 4a17 and 4b,25 as well as 1,2,4-triazines 3b,25 3c,32 3d,32 3f,33 5b,27 5c,34 5d34 are known compounds; all of these compounds were synthesized in the course of this research work and characterized by 1H, 19F, and 13C NMR spectroscopy, mass spectrometry, and elemental analysis.
Synthesis of triazines 3b–j and N-oxides 4a–j (General method I). The corresponding hydrochloride of imino ester 2 (4.0 mmol) was dissolved in solution of Na (92 mg) in MeOH (20 ml). The resulting mixture was stirred at room temperature for 1 h. The corresponding hydrazone of isonitrosoacetophenone 1 (4.0 mmol) was added and the resulting mixture was stirred at room temperature for 1 h. Solvent was removed under reduced pressure. Glacial AcOH (20 ml) was added to the residue, and the resulting mixture was refluxed for 5–10 min. Solvent was removed under reduced pressure, and the residue was purified by flash chromatography on silica gel (CHCl3 as eluent, Rf 0.7 (for 1,2,4-triazines 3b–j) and 0.3 (for 1,2,4-triazine 4-oxides 4a–j)). Analytical samples were obtained by recrystallization from MeCN.
Synthesis of triazines 3d,h and N-oxides 4d,h (General method II). Hydrochloride of imino ester 2c (743 mg, 4.0 mmol) was dissolved in MeOH (20 ml). The solution of corresponding hydrazone of isonitrosoacetophenone 1c or 1d (725 mg, 4.0 mmol) in MeOH (20 ml) was added, and the resulting mixture was stirred at room temperature overnight (for compound 1d) or for 3 days (for compound 1c). Solvent was removed under reduced pressure. Glacial AcOH (20 ml) was added to the residue, and the resulting mixture was refluxed for 5–10 min. Solvent was removed under reduced pressure, and the residue was purified by flash chromatography on silica gel (CHCl3 as eluent, Rf 0.7 (for triazines 3) and 0.3 (for N-oxides 4)). Analytical samples were obtained by recrystallization from MeCN.
Synthesis of N-oxides 4c,d,f (General method III). The corresponding hydrazone of isonitrosoacetophenone 1 (3.0 mmol) was dissolved in EtOH (30 ml), and the corresponding aldehyde (3.0 mmol) was added to solution. The resulting mixture was kept at room temperature overnight. Solvent was removed under reduced pressure, obtained residue was dissolved in AcOH (10 ml), and Pb3O4 (3058 mg, 3.0 mmol) was added by portions during 1 h by stirring at room temperature. The reaction mixture was diluted with H2O (100 ml), and the precipitate obtained was filtered off and recrystallized from EtOH.
3-Methyl-6-phenyl-1,2,4-triazin-4-ium-4-olate (4a). Yield 466 mg (62%), yellow amorphous solid, mp 181–183°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 2.72 (3H, s, CH3); 7.53–7.60 (3H, m, H Ph); 8.12–8.19 (2H, m, H Ph); 9.20 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm: 17.1; 127.4; 129.6; 131.5; 131.6; 132.7; 156.6; 159.5. Mass spectrum, m/z: 188 [M+H]+. Found, %: C 64.23; H 4.89; N 22.53. C10H9N3O. Calculated, %: C 64.16; H 4.85; N 22.45.
3-(4-Chlorophenyl)-6-phenyl-1,2,4-triazine (3b). Yield 578 mg (54%), yellow amorphous solid, mp 174–176°C. 1H NMR spectrum (CDCl3), δ, ppm: 7.51–7.56 (2H, m, C6H4Cl); 7.56–7.62 (3H, m, H Ph); 8.13–8.18 (2H, m, H Ph); 8.51–8.56 (2H, m, C6H4Cl); 9.05 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm: 126.7; 129.2; 129.4; 129.5; 131.0; 133.2; 138.1; 146.5; 155.2; 161.6. Mass spectrum, m/z: 268 [M+H]+. Found, %: C 67.48; H 3.67; N 15.73. C15H10ClN3. Calculated, %: C 67.30; H 3.77; N 15.70.
3-(4-Chlorophenyl)-6-phenyl-1,2,4-triazin-4-ium-4-olate (4b). Yield 63 mg (6%), light-yellow amorphous solid, mp 202–204°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.57–7.63 (5H, m, H Ph, C6H4Cl); 8.23–8.28 (2H, m, H Ph); 8.38–8.43 (2H, m, C6H4Cl); 9.34 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm: 127.6; 128.4; 128.8; 129.7; 129.8; 132.0; 132.4; 133.4; 136.7; 155.9; 156.3. Mass spectrum, m/z: 284 [M+H]+. Found, %: C 63.52; H 3.42; N 14.94. C15H10ClN3O. Calculated, %: C 63.50; H 3.55; N 14.81.
6-(4-Methoxyphenyl)-3-phenyl-1,2,4-triazine (3c). Yield 524 mg (50%), yellow amorphous solid, mp 168–170°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 3.90 (3H, s, OCH3); 7.10–7.15 (2H, m, C6H4OCH3); 7.55–7.60 (3H, m, H Ph); 8.21–8.26 (2H, m, C6H4OCH3); 8.47–8.53 (2H, m, H Ph); 9.36 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm: 55.5; 114.9; 125.7; 128.0; 128.1; 128.9; 131.5; 134.8; 145.9; 154.6; 151.9; 162.0. Mass spectrum, m/z: 264 [M+H]+. Found, %: C 72.78; H 5.04; N 15.92. C16H13N3O. Calculated, %: C 72.99; H 4.98; N 15.96.
6-(4-Methoxyphenyl)-3-phenyl-1,2,4-triazin-4-ium-4-olate (4c). Yield 131 mg (12%, method I), 360 mg (43%, method III), yellow amorphous solid, mp 216–218°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 3.90 (3H, s, OCH3); 7.09–7.13 (2H, m, C6H4OCH3); 7.54–7.61 (3H, m, H Ph); 8.20–8.23 (2H, m, C6H4OCH3); 8.28–8.33 (2H, m, H Ph); 9.27 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm: 56.0; 115.2; 124.6; 128.5; 129.2; 129.7; 130.1; 131.7; 132.5; 155.9; 156.1; 162.5. Mass spectrum, m/z: 280 [M+H]+. Found, %: C 68.87; H 4.74; N 14.98. C16H13N3O2. Calculated, %: C 68.81; H 4.69; N 15.05.
6-(4-Fluorophenyl)-3-phenyl-1,2,4-triazine (3d). Yield 408 mg (41%, method I), 200 mg (20%, method II), yellow amorphous solid, mp 135–137°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.34–7.42 (2H, m, C6H4F); 7.57–7.62 (3H, m, H Ph); 8.32–8.39 (2H, m, C6H4F); 8.50–8.57 (2H, m, H Ph); 9.43 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.6 (d, J = 21.8); 128.2; 128.7 (d, J = 9.2); 129.0; 129.5 (d, J = 3.2); 131.7; 134.6; 146.1; 154.2; 162.4; 164.7 (d, J = 252.8). 19F NMR spectrum (DMSO-d6), δ, ppm: –109.75 (s). Mass spectrum, m/z: 252 [M+H]+. Found, %: C 71.56; H 4.04; N 16.62. C15H10FN3. Calculated, %: C 71.70; H 4.01; N 16.72.
6-(4-Fluorophenyl)-3-phenyl-1,2,4-triazin-4-ium-4-olate (4d). Yield 273 mg (26%, method I), 530 mg (50%, method II), 401 mg (50%, method III), yellow amorphous solid, mp 223–225°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.32–7.40 (2H, m, C6H4F); 7.54–7.65 (3H, m, H Ph); 8.31–8.37 (4H, m, C6H4F, H Ph); 9.37 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 116.7 (d, J = 21.6); 128.6; 129.0 (d, J = 2.8); 129.5; 130.1 (d, J = 8.1); 130.2; 131.9; 133.3; 155.2; 158.7; 164.6 (d, J = 250.5). 19F NMR spectrum (DMSO-d6), δ, ppm: –108.88 (s). Mass spectrum, m/z: 268 [M+H]+. Found, %: C 67.37; H 3.85; N 15.83. C15H10FN3O. Calculated, %: C 67.41; H 3.77; N 15.72.
6-(4-Fluorophenyl)-3-(4-methoxyphenyl)-1,2,4-triazine (3e). Yield 530 mg (47%), light-yellow amorphous solid, mp 169–171°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 3.88 (3H, s, OCH3); 7.15–7.20 (2H, m, C6H4OCH3); 7.44–7.50 (2H, m, C6H4F); 8.30–8.36 (2H, m, C6H4F); 8.42–8.48 (2H, m, C6H4OCH3); 9.44 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 55.9; 115.1; 116.8 (d, J = 21.1); 127.2; 129.5 (d, J = 8.7); 129.9; 130.3 (d, J = 2.8); 147.9; 153.9; 161.7; 162.7; 164.3 (d, J = 249.2). 19F NMR spectrum (DMSO-d6), δ, ppm: –110.29 (s). Mass spectrum, m/z: 282 [M+H]+. Found, %: C 68.28; H 4.23; N 14.81. C16H12FN3O. Calculated, %: C 68.32; H 4.30; N 14.94.
6-(4-Fluorophenyl)-3-(4-methoxyphenyl)-1,2,4-triazin-4-ium-4-olate (4e). Yield 134 mg (11%), yellow amorphous solid, mp 234–236°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 3.88 (3H, s, OCH3); 7.12–7.16 (2H, m, C6H4OCH3); 7.43–7.48 (2H, m, C6H4F); 8.27–8.31 (2H, m, C6H4F); 8.35–8.39 (2H, m, C6H4OCH3); 9.33 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 56.0; 114.1; 116.7 (d, J = 21.5); 121.6; 129.1 (d, J = 3.3); 130.7 (d, J = 9.1); 132.0; 133.3; 154.6; 156.1; 162.4; 164.6 (d, J = 249.7). 19F NMR spectrum (DMSO-d6), δ, ppm: –109.43 (s). Mass spectrum, m/z: 298 [M+H]+. Found, %: C 64.78; H 4.02; N 14.01. C16H12FN3O. Calculated, %: C 64.64; H 4.07; N 14.13.
6-(4-Fluorophenyl)-3-(thiophen-2-yl)-1,2,4-triazine (3f). Yield 165 mg (16%), yellow amorphous solid, mp 154– 156°C 1H NMR spectrum (DMSO-d6), δ, ppm: 7.25–7.30 (1H, m, H thienyl); 7.33–7.40 (2H, m, C6H4F); 7.78–7.82 (1H, m, H thienyl); 8.10–8.14 (1H, m, H thienyl); 8.27–8.34 (2H, m, C6H4F); 9.34 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.6 (d, J = 21.8); 128.5 (d, J = 8.7); 128.7; 129.5 (d, J = 3.7); 130.3; 131.4; 139.4; 146.1; 153.6; 159.9; 164.6 (d, J = 252.6). 19F NMR spectrum (DMSO-d6), δ, ppm: –109.84 (s). Mass spectrum, m/z: 258 [M+H]+. Found, %: C 60.77; H 3.23; N 16.45. C13H8FN3S. Calculated, %: C 60.69, H 3.13, N 16.33.
6-(4-Fluorophenyl)-3-(thiophen-2-yl)-1,2,4-triazin-4-ium-4-olate (4f). Yield 440 mg (40%, method I), 440 mg (54%, method III), yellow amorphous solid, mp 193–195°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.31–7.40 (3H, m, H thienyl, C6H4F); 7.90–7.95 (1H, m, H thienyl); 8.28–8.35 (2H, m, C6H4F); 8.58–8.62 (1H, m, H thienyl); 9.43 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 116.7 (d, J = 21.5); 128.1; 129.1 (d, J = 3.2); 129.1; 129.9 (d, J = 9.0); 132.2; 132.7; 134.5; 152.8; 153.5; 164.5 (d, J = 249.7). 19F NMR spectrum (DMSO-d6), δ, ppm: –109.19 (s). Mass spectrum, m/z: 274 [M+H]+. Found, %: C 57.08; H 3.01; N 15.51. C13H8FN3OS. Calculated, %: C 57.13; H 2.95; N 15.38.
3-(4-Chlorophenyl)-6-(4-fluorophenyl)-1,2,4-triazine (3g). Yield 612 mg (54%), yellow amorphous solid, mp 170– 172°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.33–7.41 (2H, m, C6H4F); 7.58–7.63 (2H, m, C6H4Cl); 8.32–8.38 (2H, m, C6H4F); 8.49–8.54 (2H, m, C6H4Cl); 9.44 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.6 (d, J = 22.2); 128.7 (d, J = 8.1); 129.2; 129.4 (d, J = 3.1); 129.5; 133.1; 138.2; 146.0; 154.3; 161.6; 164.8 (d, J = 252.3). 19F NMR spectrum (DMSO-d6), δ, ppm: –109.62 (s). Mass spectrum, m/z: 286 [M+H]+. Found, %: C 63.16; H 3.24; N 14.81. C15H9ClFN3. Calculated, %: C 63.06; H 3.18; N 14.71.
3-(4-Chlorophenyl)-6-(4-fluorophenyl)-1,2,4-triazin-4-ium-4-olate (4g). Yield 121 mg (10%), yellow amorphous solid, mp 211–213°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.32–7.41 (2H, m, C6H4F); 7.57–7.64 (2H, m, C6H4Cl); 8.29–8.36 (2H, m, C6H4F); 8.36–8.42 (2H, m, C6H4Cl); 9.38 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 116.8 (d, J = 22.3); 128.4; 128.8; 129.0 (d, J = 3.3); 130.2 (d, J = 7.9); 132.0; 133.4; 136.8; 155.5; 156.8; 164.6 (d, J = 247.9). 19F NMR spectrum (DMSO-d6), δ, ppm: –108.75 (s). Mass spectrum, m/z: 302 [M+H]+. Found, %: C 59.84, H 3.12, N 13.79. C15H9ClFN3O. Calculated, %: C 59.71; H 3.01; N 13.93.
6-(3-Fluorophenyl)-3-phenyl-1,2,4-triazine (3h). Yield 559 mg (56%, method I), 309 mg (31%, method II), yellow amorphous solid, mp 178–180°C. 1H NMR spectrum (CDCl3), δ, ppm: 7.24–7.28 (1H, m, C6H4F); 7.54–7.60 (4H, m, C6H4F, H Ph); 7.91–7.95 (2H, m, C6H4F); 8.58–8.61 (2H, m, H Ph); 9.07 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 113.7 (d, J = 23.3); 117.8 (d, J = 21.2); 122.2 (d, J = 3.0); 128.3; 128.9; 131.0 (d, J = 8.1); 131.8; 134.5; 135.6 (d, J = 8.0); 146.3; 154.0 (d, J = 3.0); 162.8; 163.5 (d, J = 249.1). 19F NMR spectrum (CDCl3), δ, ppm: –111.02 (s). Mass spectrum, m/z: 252 [M+H]+. Found, %: C 71.57; H 4.06; N 16.79. C15H10FN3. Calculated, %: C 71.70; H 4.01; N 16.72.
6-(3-Fluorophenyl)-3-phenyl-1,2,4-triazin-4-ium-4-olate (4h). Yield 59 mg (6%, method I), 329 mg (31%, method II), yellow amorphous solid, mp 208–210°C. 1H NMR spectrum (CDCl3), δ, ppm: 7.27–7.32 (1H, m, C6H4F); 7.54–7.58 (3H, m, H Ph); 7.59–7.62 (1H, m, C6H4F); 7.79–7.82 (1H, m, C6H4F); 7.84–7.87 (1H, m, C6H4F); 8.43–8.47 (2H, m, H Ph); 8.62 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 114.1 (d, J = 23.7); 118.7 (d, J = 20.9); 122.5 (d, J = 3.8); 128.3; 128.4; 130.0; 131.2 (d, J = 8.9); 132.2 (2C); 133.9 (d, J = 8.2); 154.9 (d, J = 3.0); 157.0; 163.4 (d, J = 248.8). 19F NMR spectrum (CDCl3), δ, ppm: –111.53 (s). Mass spectrum, m/z: 268 [M+H]+. Found, %: C 67.53; H 3.89; N 15.61. C15H10FN3O. Calculated, %: C 67.41; H 3.77; N 15.72.
6-(3-Fluorophenyl)-3-(4-methoxyphenyl)-1,2,4-triazine (3i). Yield 450 mg (40%), yellow amorphous solid, mp 197– 199°C. 1H NMR spectrum (CDCl3), δ, ppm: 3.92 (3H, s, OCH3); 7.04–7.10 (2H, m, C6H4OCH3); 7.21–7.25 (1H, m, C6H4F); 7.54–7.58 (1H, m, C6H4F); 7.88–7.93 (2H, m, C6H4F); 8.53–8.58 (2H, m, C6H4OCH3); 8.99 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 55.9; 113.7 (d, J = 23.9); 115.1; 118.0 (d, J = 20.8); 123.1 (d, J = 3.0); 127.1; 130.0; 131.8 (d, J = 8.0); 136.1 (d, J = 8.0); 148.2; 153.5 (d, J = 3.0); 162.0; 162.8; 163.2 (d, J = 244.2). 19F NMR spectrum (CDCl3), δ, ppm: –111.16 (s). Mass spectrum, m/z: 282 [M+H]+. Found, %: C 68.48; H 4.27; N 15.03. C16H12FN3O. Calculated, %: C 68.32; H 4.30; N 14.94.
6-(3-Fluorophenyl)-3-(4-methoxyphenyl)-1,2,4-triazin-4-ium-4-olate (4i). Yield 202 mg (17%), yellow amorphous solid, mp 226–228°C. 1H NMR spectrum (CDCl3), δ, ppm: 3.92 (3H, s, OCH3); 7.03–7.09 (2H, m, C6H4OCH3); 7.26–7.32 (1H, m, C6H4F); 7.51–7.59 (1H, m, C6H4F); 7.76–7.81 (1H, m, C6H4F); 7.81–7.87 (1H, m, C6H4F); 8.56–8.60 (3H, m, C6H4OCH3, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 55.9; 114.1; 114.2 (d, J = 22.3); 118.5 (d, J = 22.3); 121.5; 123.6 (d, J = 3.0); 130.0; 131.8 (d, J = 8.2); 132.1; 133.7; 134.9 (d, J = 8.2); 154.2 (d, J = 3.0); 156.5; 163.1 (d, J = 244.0). 19F NMR spectrum (CDCl3), δ, ppm: –110.65 (s). Mass spectrum, m/z: 298 [M+H]+. Found, %: C 64.59; H 4.11; N 14.03. C16H12FN3O2. Calculated, %: C 64.64; H 4.07; N 14.13.
6-(3-Fluorophenyl)-3-(thiophen-2-yl)-1,2,4-triazine (3j). Yield 219 mg (21%), yellow amorphous solid, mp 166– 168°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.26–7.30 (1H, m, H thienyl); 7.32–7.38 (1H, m, C6H4F); 7.60–7.67 (1H, m, C6H4F); 7.82–7.85 (1H, m, H thienyl); 8.03–8.11 (2H, m, C6H4F); 8.13–8.16 (1H, m, H thienyl); 9.38 (1H, s, H-5). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 113.8 (d, J = 23.5); 118.1 (d, J = 21.5); 123.2 (d, J = 3.0); 129.6; 130.8; 131.9 (d, J = 8.1); 133.0; 136.0 (d, J = 8.1); 139.4; 148.5; 153.7 (d, J = 3.0); 159.9; 163.2 (d, J = 244.8). 19F NMR spectrum (DMSO-d6), δ, ppm: –111.58 (s). Mass spectrum, m/z: 258 [M+H]+. Found, %: C 60.79; H 3.11; N 16.42. C13H8FN3S. Calculated, %: C 60.69; H 3.13; N 16.33.
6-(3-Fluorophenyl)-3-(thiophen-2-yl)-1,2,4-triazin-4-ium-4-olate (4j). Yield 470 mg (43%), yellow amorphous solid, mp 191–193°C. 1H NMR spectrum (DMSO-d6), δ, ppm: 7.32–7.40 (2H, m, H thienyl, C6H4F); 7.58–7.66 (1H, m, C6H4F); 7.92–7.97 (1H, m, H thienyl); 8.03–8.13 (2H, m, C6H4F, H thienyl); 8.59–8.63 (1H, m, H thienyl); 9.48 (1H, s, H-5). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 113.9 (d, J = 21.3); 118.5 (d, J = 21.3); 122.2 (d, J = 3.0): 127.7; 128.5; 130.7; 131.2 (d, J = 8.0); 133.4; 134.0 (d, J = 8.0); 134.1; 153.1 (d, J = 3.0); 153.5; 163.4 (d, J = 248.9). 19F NMR spectrum (DMSO-d6), δ, ppm: –111.57 (s). Mass spectrum, m/z: 274 [M+H]+. Found, %: C 57.20; H 2.92; N 15.23. C13H8FN3OS. Calculated, %: C 57.13; H 2.95; N 15.38.
Synthesis of pyridines 5b–j (General method IV). The corresponding triazine 3 (0.6 mmol) was suspended in o-xylene (for triazines 3d,e,h,j) or in 1,2-dichlorobenzene (for triazines 3b,c,f,g,i) (25 ml), 2,5-norbornadiene (0.30 ml, 3.0 mmol) was added, and the resulting mixture was refluxed for 36 h with addition of 2,5-norbornadiene (0.15 ml, 1.5 mmol) every 12 h. Solvent was removed under reduced pressure. The product was isolated by column chromatography on silica gel (CHCl3, Rf 0.6). Solvent from fractions containing product was removed under reduced pressure. The residue was recrystallized from EtOH.
2-(4-Chlorophenyl)-5-phenylpyridine (5b). Yield 101 mg (63%), light-yellow crystalline solid, mp 177–179. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.40–7.46 (1H, m, H Ph); 7.48–7.55 (4H, m, H Ph, C6H4Cl); 7.72–7.77 (2H, m, H Ph); 8.02 (1H, d, 3J = 8.4, H-3); 8.10–8.18 (3H, m, C6H4Cl, H-4); 8.94 (1H, d, 4J = 2.0, H-6). 13C NMR spectrum (CDCl3), δ, ppm: 120.2; 127.0; 128.1; 128.2; 129.0; 129.2; 135.2; 135.2; 135.3; 137.4; 137.5; 148.1; 154.9. Mass spectrum, m/z: 266 [M+H]+. Found, %: C 76.97; H 4.43; N 5.12. C17H12ClN. Calculated, %: C 76.84; H 4.55; N 5.27.
5-(4-Methoxyphenyl)-2-phenylpyridine (5c). Yield 102 mg (65%), yellow amorphous solid, mp 207–208°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.87 (3H, s, CH3O); 7.00–7.06 (2H, m, C6H4OCH3); 7.39–7.45 (1H, m, H Ph); 7.46–7.53 (2H, m, H Ph); 7.55–7.61 (2H, m, H Ph); 7.79 (1H, d, 3J = 8.0, H-3); 7.91 (1H, dd, 3J = 8.0, 4J = 2.4, H-4); 8.01–8.07 (2H, m, C6H4OCH3); 8.91 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (CDCl3), δ, ppm: 55.4; 114.6; 120.3; 126.8; 128.1; 128.8; 128.9; 130.1; 134.6; 139.1; 147.7; 155.6; 159.8. Mass spectrum, m/z: 262 [M+H]+. Found, %: C 82.61; H 5.85; N 5.24. C18H15NO. Calculated, %: C 82.73; H 5.79; N 5.36.
5-(4-Fluorophenyl)-2-phenylpyridine (5d). Yield 95 mg (63%), light-grey amorphous solid, mp 178–180°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.16–7.22 (2H, m, C6H4F); 7.40–7.46 (1H, m, Ph); 7.47–7.53 (2H, m, H Ph); 7.57–7.63 (2H, m, C6H4F); 7.81 (1H, d, 3J = 8.0, H-3); 7.91 (1H, dd, 3J = 8.0, 4J = 2.4, H-4); 8.02–8.07 (2H, m, H Ph); 8.93 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.1 (d, J = 21.1); 120.5; 126.9; 128.7 (d, J = 8.1); 128.9; 129.1; 133.7 (d, J = 3.1); 134.0; 135.1; 138.8; 147.8; 156.2; 163.0 (d, J = 248.4). 19F NMR spectrum (DMSO-d6), δ, ppm: –114.24 (s). Mass spectrum, m/z: 250 [M+H]+. Found, %: C 81.83; H 4.94; N 5.69. C17H12FN. Calculated, %: C 81.91; H 4.85; N 5.62.
5-(4-Fluorophenyl)-2-(4-methoxyphenyl)pyridine (5e). Yield 109 mg (65%), yellow amorphous solid, mp 189– 191°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.88 (3H, s, OCH3); 6.99–8.05 (2H, m, C6H4OCH3); 7.15–7.21 (2H, m, C6H4F); 7.55–7.61 (2H, m, C6H4F); 7.74 (1H, d, 3J = 8.0, H-3); 7.88 (1H, dd, 3J = 8.0, 4J = 2.4, H-4); 7.97–8.03 (2H, m, C6H4OCH3); 8.85 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 55.7; 114.7; 116.5 (J = 21.8); 119.7; 128.3; 129.2 (J = 8.2); 131.1; 132.8; 133.9 (J = 3.1); 135.4; 147.8; 155.1; 160.7; 162.7 (J = 245.6). 19F NMR spectrum (CDCl3), δ, ppm: –114.43 (s). Mass spectrum, m/z: 280 [M+H]+. Found, %: C 77.26; H 4.92; N 5.11. C18H14FNO. Calculated, %: C 77.40; H 5.05; N 5.01.
5-(4-Fluorophenyl)-2-(thiophen-2-yl)pyridine (5f). Yield 94 mg (61%), light-yellow crystalline solid, mp 169–171°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.12–7.21 (3H, m, H thienyl, C6H4F); 7.40–7.44 (1H, m, H thienyl); 7.53–7.60 (2H, m, C6H4F); 7.60–7.64 (1H, m, H thienyl); 7.72 (1H, d, 3J = 8.0, H-3); 7.84 (1H, dd, 3J = 8.0, 4J = 2.0, H-4); 8.77 (1H, d, 4J = 2.0, H-6). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.1 (d, J = 21.3); 118.7; 124.7; 127.8; 128.2; 128.5 (d, J = 8.0); 133.6 (d, J = 3.7); 133.7; 134.8; 144.5; 147.7; 151.4; 162.9 (d, J = 248.2). 19F NMR spectrum (CDCl3), δ, ppm: –114.17 (s). Mass spectrum, m/z: 256 [M+H]+. Found, %: C 70.51; H 4.08; N 5.34. C15H10FNS. Calculated, %: C 70.57; H 3.95; N 5.49.
2-(4-Chlorophenyl)-5-(4-fluorophenyl)pyridine (5g). Yield 116 mg (68%), light-yellow crystalline solid mp 173– 175°C. 1H NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 7.25–7.32 (2H, m, C6H4F); 7.47–7.53 (2H, m, C6H4Cl); 7.75–7.82 (2H, m, C6H4F); 8.02 (1H, d, 3J = 8.4, H-3); 8.11 (1H, dd, 3J = 8.4, 4J = 2.4, H-4); 8.12–8.17 (2H, m, C6H4Cl); 8.92 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 116.2 (d, J = 21.6); 120.2; 128.1; 128.7 (d, J = 8.0); 129.0; 133.3 (d, J = 3.1); 134.3; 135.1; 135.2; 137.3; 148.0; 154.9; 163.0 (d, J = 248.2). 19F NMR spectrum (DMSO-d6), δ, ppm: –114.12 (s). Mass spectrum, m/z: 284 [M+H]+. Found, %: C 72.08; H 4.02; N 4.81. C17H11ClFN. Calculated, %: C 71.96; H 3.91; N 4.94.
5-(3-Fluorophenyl)-2-phenylpyridine (5h). Yield 92 mg (61%), yellow amorphous solid, mp 186–188°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.08–7.14 (1H, m, C6H4F); 7.31–7.36 (1H, m, C6H4F); 7.40–7.54 (5H, m, C6H4F, H Ph); 7.83 (1H, d, 3J = 8.0, H-3); 7.94 (1H, dd, 3J = 8.0, 4J = 2.4, H-4); 8.03–8.08 (2H, m, H Ph); 8.92 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (DMSO-d6), δ, ppm (J, Hz): 114.0 (d, J = 22.6); 115.4 (d, J = 21.6); 120.6; 123.3 (d, J = 2.1); 127.0; 129.3; 129.7; 131.6 (d, J = 8.5); 133.3 (d, J = 2.1); 135.8; 138.6; 139.7 (d, J = 7.7); 148.2; 155.8; 163.3 (d, J = 244.1). 19F NMR spectrum (CDCl3), δ, ppm: –112.25 (s). Mass spectrum, m/z: 250 [M+H]+. Found, %: C 82.79; H 4.78; N 5.64. C17H12FN. Calculated, %: C 81.91; H 4.85; N 5.62.
5-(3-Fluorophenyl)-2-(4-methoxyphenyl)pyridine (5i). Yield 103 mg (61%), yellow amorphous solid, mp 192– 194°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 3.89 (3H, s, OCH3); 7.00–7.05 (2H, m, C6H4OCH3); 7.07–7.13 (1H, m, C6H4F); 7.30–7.35 (1H, m, C6H4F); 7.39–7.49 (2H, m, C6H4F); 7.78 (1H, d, 3J = 8.0, H-3); 7.93 (1H, dd, 3J = 8.0, 4J = 2.4, H-4); 8.00–8.05 (2H, m, C6H4OCH3); 8.89 (1H, d, 4J = 2.4, H-6). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 55.4; 113.8 (d, J = 22.8); 114.3; 114.8 (d, J = 21.7); 119.7; 122.6 (d, J = 3.0); 128.3; 130.7 (d, J = 8.4); 131.1; 133.0 (d, J = 2.2); 135.3; 139.9 (d, J = 8.4); 147.6; 156.3; 160.8; 163.3 (d, J = 247.2). 19F NMR spectrum (CDCl3), δ, ppm: –49.50 (s). Mass spectrum, m/z: 280 [M+H]+. Found, %: C 77.51; H 5.17; N 5.13. C18H14FNO. Calculated, %: C 77.40; H 5.05; N 5.01.
5-(3-Fluorophenyl)-2-(thiophen-2-yl)pyridine (5j). Yield 98 mg (64%), yellow crystalline solid, mp 176–178°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 7.06–7.16 (2H, m, H thienyl, C6H4F); 7.28–7.33 (1H, m, C6H4F); 7.36–7.41 (1H, m, C6H4F); 7.41–7.49 (2H, m, H thienyl, C6H4F); 7.61–7.65 (1H, m, H thienyl); 7.73 (1H, d, 3J = 8.0, H-3); 7.87 (1H, dd, 3J = 8.0, 4J = 2.0, H-4); 8.80 (1H, d, 4J = 2.0, H-6). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 113.7 (d, J = 21.9); 114.9 (d, J = 21.2); 116.7; 122.5 (d, J = 3.1); 124.9; 127.9; 128.2; 130.7 (d, J = 8.8); 133.4 (d, J = 2.2); 134.9; 139.8 (d, J = 7.2); 144.4; 147.8; 152.0; 163.3 (d, J = 246.5). 19F NMR spectrum (CDCl3), δ, ppm: –112.18 (s). Mass spectrum, m/z: 256 [M+H]+. Found, %: C 70.63; H 3.87; N 5.54. C15H10FNS. Calculated, %: C 70.57; H 3.95; N 5.49.
Synthesis of pyridines 6c,f (General method V). The mixture of the corresponding triazine 3c or 3f (0.5 mmol) and 1-morpholinocyclopentene (0.40 ml, 2.5 mmol) was stirred at 200°C for 2 h under argon atmosphere. Then the additional portion of 1-morpholinocyclopentene (0.20 ml, 1.25 mmol) was added and the resulting mixture was stirred for additional 1 h at the same conditions. The reaction mass was cooled to room temperature. The products were separated by flash chromatography on silica gel (DCM as eluent, Rf 0.6) and then were purified by recrystallization (EtOH).
4-(4-Methoxyphenyl)-1-phenyl-5H,6H,7H-cyclopenta-[c]pyridine (6c). Yield 62 mg (41%), light-grey crystalline solid, mp 169–171°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.01–2.12 (2H, m, 6-CH2); 3.05 (2H, t, 3J = 7.6, 7-CH2); 3.17 (2H, t, 3J = 7.6, 5-CH2); 3.88 (3H, s, OCH3); 6.99–7.04 (2H, m, C6H4OCH3); 7.37–7.51 (5H, m, H Ph); 7.81–7.82 (2H, m, C6H4OCH3); 8.54 (1H, s, H-3). 13C NMR spectrum (DMSO-d6), δ, ppm: 25.9; 32.9; 33.1; 55.7; 114.7; 128.6; 128.7; 128.7; 129.8; 130.1; 132.2; 137.6; 140.1; 146.9; 151.9; 152.4; 159.4. Mass spectrum, m/z: 302 [M+H]+. Found, %: C 83.82; H 6.48; N 4.52. C21H19NO. Calculated, %: C 83.69; H 6.35; N 4.65.
4-(4-Fluorophenyl)-1-(thiophen-2-yl)-5H,6H,7H-cyclo-penta[c]pyridine (6f). Yield 71 mg (48%), light-grey crystalline solid, mp 155–157°C. 1H NMR spectrum (CDCl3), δ, ppm (J, Hz): 2.21–2.22 (2H, m, 6-CH2); 3.02 (2H, t, 3J = 7.6, 7-CH2); 3.26 (2H, t, 3J = 7.6, 5-CH2); 7.12–7.19 (3H, m, H thienyl, C6H4F); 7.39–7.45 (3H, m, H thienyl, C6H4F); 7.52–7.55 (1H, m, H thienyl); 8.42 (1H, s, H-3). 13C NMR spectrum (CDCl3), δ, ppm (J, Hz): 25.0; 32.6; 33.2; 115.7 (d, J = 21.9); 126.2; 127.4; 128.0; 130.1 (d, J = 8.4); 131.4; 133.8 (d, J = 3.0); 135.3; 145.1; 146.7; 147.3; 152.7; 162.5 (d, J = 247.6). 19F NMR spectrum (CDCl3), δ, ppm: –114.41 (s). Mass spectrum, m/z: 296 [M+H]+. Found, %: C 73.32; H 4.89; N 4.61. C18H14FNS. Calculated, %: C 73.19; H 4.78; N 4.74.
The X-ray diffraction analyses of compounds 3b, 4e, and 5g were carried out using equipment of the Center for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis of the Russian Academy of Sciences (Ural Branch). The experiments were accomplished on the automated X-ray diffractometer Xcalibur 3 with CCD detector on standard procedure (MoKα irradiation, graphite monochromator, ω-scans with 1° step at 295(2)K). Empirical absorption correction was applied. The solution and refinement of the structures were accomplished using the Olex2 program package.35 The structures were solved by direct method in the ShelXS program and refined by the ShelXL by full-matrix least-squared method in anisotropic approximation for non-hydrogen atoms.36 The H atoms were placed in the calculated positions and were refined in the isotropic approximation.
Compound 3b. Crystal data for C15H10ClN3 (M 267.71 g/mol): monoclinic, space group P21/n; a 11.8295(10), b 7.6753(7), c 27.459(2) Å; β 95.589(7)°; V 2481.3(4) Å3; Z 8, T 295(2)K, μ(MoKα) 0.295 mm–1; Dcalc 1.433 g/cm3. 16864 Reflections measured (2.76° ≤ Θ ≤ 29.14°), 6240 unique (Rint 0.0478, Rsigma 0.0942) and 2391 reflections with I > 2σ(I) which were used in all calculations. The final R1 0.0488 (I > 2σ(I)) and wR2 0.0804 (all data), GOOF 1.007. Largest difference peak/hole 0.240/–0.250 ēÅ–3.
Compound 4e. Crystal data for C16H12FN3O2 (M 297.29 g/mol): monoclinic, space group P21/n; a 12.4029(16), b 6.5008(5), c 17.2879(17) Å; β 106.773(12)°; V 1334.6(2) Å3; Z 4; T 295(2)K; μ(MoKα) 0.110 mm–1; Dcalc 1.480 g/cm3. 5202 Reflections measured (4.76° ≤ Θ ≤ 54.2°), 2915 unique (Rint 0.0515, Rsigma 0.1421) and 1141 reflections with I > 2σ(I) which were used in all calculations. The final R1 0.0613 (I > 2σ(I)) and wR2 0.1265 (all data), GOOF 0.964. Largest difference peak/hole 0.17/–0.20 ēÅ–3.
Compound 5g. Crystal data for C17H11ClFN (M 283.72 g/mol): orthorhombic, space group Pbca; a 10.0665(12), b 10.1880(12), c 26.244(4) Å; β 90°; V 2691.6(6) Å3; Z 8; T 295(2)K; μ(MoKα) 0.283 mm–1; Dcalc 1.400 g/cm3. 14676 Reflections measured (7.354° ≤ Θ ≤ 56.564°), 3280 unique (Rint 0.0571, Rsigma 0.0407) and 1943 reflections with I > 2σ(I) which were used in all calculations. The final R1 0.0574 (I > 2σ(I)) and wR2 0.1974 (all data), GOOF 1.043. Largest difference peak/hole 0.19/–0.29 ēÅ–3.
The final atomic coordinates and crystallographic data for compounds 3b, 4e, and 5g have been deposited at the Cambridge Crystallographic Data Center (deposits CCDC 2298502, CCDC 2309894, and CCDC 2309892, respectively).
Supplementary information file containing 1H, 13C, and 19F NMR spectra of compounds 3b–j, 4a–j, 5b–j, 6c,f, the selected bond distances and angles for compound 3b, and additional photophysical data for compounds 6c,f, is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
This work was supported by the Russian Science Foundation grant # 19-73-10144-P.
References
Chen, Y.; Rosenkranz, C.; Hirte, S.; Kirchmair, J. Nat. Prod. Rep. 2022, 39, 1544.
De, S.; Kumar S. K. A.; Shah, S. K.; Kazi, S.; Sarkar, N.; Banerjee, S.; Dey, S. RSC Adv. 2022, 12, 15385.
Kutlu, E.; Emen, F. M.; Kismali, G.; Kınaytürk, N. K.; Kılıç, D.; Karacolak, A. I.; Demirdogen, R. E. J. Mol. Struct. 2021, 1234, 130191.
Xu, M.; Li, W.; An, Z.; Zhou, Q.; Wang, G. Appl. Organomet. Chem. 2005, 19, 1225.
Li, Y.; Xu, K.; Wen, X.; Zhang, L.; Yin, Y.; Liu, S.; Piao, X.; Xie, W. Org. Electron. 2013, 14, 1946.
Ishida, N.; Moriya, T.; Goya, T.; Murakami, M. J. Org. Chem. 2010, 75, 8709.
Liu, K.; Lalancette, R. A.; Jäkle, F. J. Am. Chem. Soc. 2017, 139, 18170.
Cai, Z.; Zhang, W.; Cao, Y.; Du, X. J. Heterocycl. Chem. 2022, 59, 1247.
Cao, Y.-Y.; Mao, D.-J.; Wang, W.-W.; Du, X.-H J. Agric. Food Chem. 2017, 65, 6114.
Jacquemard, U.; Routier, S.; Dias, N.; Lansiaux, A.; Goossens, J.-F.; Bailly, C.; Mérour, J.-Y. Eur. J. Med. Chem. 2005, 40, 1087.
Pabst, G. R.; Pfüller, O. C.; Sauer, J. Tetrahedron 1999, 55, 8045.
Taylor, E. C.; Macor, J. E. J. Org. Chem. 1989, 54, 1249.
Prokhorov, A. M.; Kozhevnikov, D. N. Chem. Heterocycl. Compd. 2012, 48, 1153.
Fatykhov, R. F.; Sharapov, A. D.; Starnovskaya, E. S.; Shtaitz, Y. K.; Savchuk, M. I.; Kopchuk, D. S.; Nikonov, I. L.; Zyryanov, G. V.; Khalymbadzha, I. A.; Chupakhin, O. N. Spectrochim. Acta, Part A 2022, 267, 120499.
Santoro, A.; Whitwood, A. C.; Williams, J. A. G.; Kozhevnikov, V. N.; Bruce, D. W. Chem. Mater. 2009, 21, 3871.
Khasanov, A. F.; Kopchuk, D. S.; Kim, G. A.; Slepukhin, P. A.; Kovalev, I. S.; Santra, S.; Zyryanov, G. V.; Majee, A.; Chupakhin, O. N.; Charushin, V. N. ChemistrySelect 2018, 3, 340.
Kozhevnikov, D. N.; Kozhevnikov, V. N.; Rusinov, V. L.; Chupakhin, O. N. Mendeleev Commun. 1997, 7, 238.
Kozhevnikov, V. N.; Kozhevnikov, D. N.; Shabunina, O. V.; Rusinov, V. L.; Chupakhin, O. N. Tetrahedron Lett. 2005, 46, 1791.
Bennett, G. B.; Mason, R. B.; Alden, L. J.; Roach, J. B. J. Med. Chem. 1978, 21, 623.
Kozhevnikov, D. N.; Kozhevnikov, V. N.; Rusinov, V. L.; Chupakhin, O. N. Chem. Heterocycl. Compd. 1999, 35, 1377.
Kozhevnikov, D. N.; Kataeva, N. N.; Rusinov, V. L.; Chupakhin, O. N. Russ. Chem. Bull. 2004, 53, 1295.
Kozhevnikov, V. N.; Shabunina, O. V.; Kopchuk, D. S.; Ustinova, M. M.; König, B.; Kozhevnikov, D. N. Tetrahedron 2008, 64, 8963.
Böhnisch, V.; Burzer, G.; Neunhoeffer, H. Justus Liebigs Ann. Chem. 1977, 1713.
Hunter, M. J.; Ludwig, M. L. J. Am. Chem. Soc. 1962, 84, 3491.
Crespin, L.; Biancalana, L.; Morack, T.; Blakemore, D. C.; Ley, S. V. Org. Lett. 2017, 19, 1084.
Kozhevnikov, V. N.; Ustinova, M. M.; Slepukhin, P. A.; Santoro, A.; Bruce, D. W.; Kozhevnikov, D. N. Tetrahedron Lett. 2008, 49, 4096.
Wang, S.-W.; Guo, W.-S.; Wen, L.-R.; Li, M. RSC Adv. 2014, 4, 59218.
Kozhevnikov, D. N.; Shabunina, O. V.; Kopchuk, D. S.; Slepukhin, P. A.; Kozhevnikov, V. N. Tetrahedron Lett. 2006, 47, 7025.
Guda, M. R.; Valieva, M. I.; Kopchuk, D. S.; Aluru, R.; Khasanov, A. F.; Taniya, O. S.; Novikov, A. S.; Zyryanov, G. V.; Ranu, B. C. J. Fluoresc. 2024, 34, 579.
Kopchuk, D. S.; Chepchugov, N. V.; Starnovskaya, E. S.; Khasanov, A. F.; Krinochkin, A. P.; Santra, S.; Zyryanov, G. V.; Das, P.; Majee, A.; Rusinov, V. L.; Charushin, V. N. Dyes Pigm. 2019, 167, 151.
Porrès, L.; Holland, A.; Pålsson, L.-O.; Monkman, A. P.; Kemp, C.; Beeby, A. J. Fluoresc. 2006, 16, 267.
Meng, J.; Wen, M.; Zhang, S.; Pan, P.; Yu, X.; Deng, W.-P. J. Org. Chem. 2017, 82, 1676.
Shafikov, M. Z.; Kozhevnikov, D. N.; Bodensteiner, M.; Brandl, F.; Czerwieniec, R. Inorg. Chem. 2016, 55, 7457.
Sivakumar, G.; Subaramanian, M.; Balaraman, E. ACS Sustainable Chem. Eng. 2022, 10, 7362.
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339.
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, A71, 3.
Author information
Authors and Affiliations
Corresponding author
Additional information
In memory of Dr. Prof. H. Neunhoeffer (1936–2018)
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2024, 60(5/6), 289–298
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Krinochkin, A.P., Khasanov, A.F., Valieva, M.I. et al. 2,5-Di(het)arylpyridines: synthesis by ‘‘1,2,4-triazine’’ methodology and photophysical properties. Chem Heterocycl Comp 60, 289–298 (2024). https://doi.org/10.1007/s10593-024-03335-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-024-03335-x