
Abstract
Purpose
Primary immunodeficiency diseases (PIDs) are inherited disorders of the immune system resulting in increased susceptibility to unusual infections and predisposition to autoimmunity and malignancies. The European Society for Immunodeficiencies (ESID) has developed an internet-based database for clinical and research data on patients with PID. This study aimed to provide a minimum estimate of the prevalence of each disorder and to determine the clinical characteristics and outcomes of patients with PID in Turkey.
Methods
Clinical features of 1435 patients with primary immunodeficiency disorders are registered in ESID Online Patient Registry by the Pediatric Immunology Departments of the Medical Faculties of Uludag University and Ege University Between 2004 and 2010. These two centers are the major contributors reporting PID patients to ESID database from Turkey.
Results
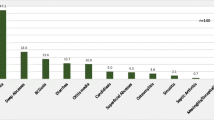
Predominantly antibody immunodeficiency (73.5 %) was the most common category followed by autoinflammatory disorders (13.3 %), other well defined immunodeficiencies (5.5 %), congenital defects of phagocyte number, function or both 3.5 %), combined T and B cell immunodeficiencies (2 %), defects in innate immunity (1 %), and diseases of immune dysregulation (0.7 %). Patients between 0 and 18 years of age constitued 94 % of total and the mean age was 9.2 ± 6 years. The consanguinity rate within the registered patients was 14.3 % (188 of 1130 patients). The prevalance of all PID cases ascertained from the registry was 30.5/100.000. The major cause of the mortality was severe infection which was seen in forty-two of seventy five deceased patients. The highest mortality was observed in patients with severe combined immunodeficiencies and ataxia-telangiectasia.
Conclusion
Promoting the awareness of PID among the medical professionals and the general public is required if premature death and serious morbidity occurs due to late diagnosis of the wider spectrum of PID are to be avoided.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiency diseases (PID) are a heterogeneous group of inherited disorders of the immune system, predisposing individuals to recurrent infections, allergy, autoimmunity, and malignancies. The phenotypic diversity of PIDs is contributed by the heterogeneity of mutations that may affect single genes and that may variably affect protein expression and function. Clinical descriptions have already been made for more than 200 PIDs [1–3], for which over 150 forms of PID have been molecularly characterized and this number is still expanding.
The International Union of Immunological Societies (IUIS) classifies diseases into 8 major groups and subgroups [4]: combined T- and B-cell immunodeficiencies, predominantly antibody deficiencies, other well-defined immunodeficiency syndromes, diseases of immune dysregulation, congenital defects of phagocyte number, function, or both, defects in innate immunity, autoinflammatory disorders, and complement deficiencies.
Epidemiological studies have shown wide geographical and racial variations in the prevalence and the pattern of immunodeficiency diseases. Several PID patient registries have been released in different countries [5–15]. These registries helped in determining the frequency of PID in these countries. The ESID patient registry is the largest and a secure, internet based patient registry, which combines both clinical and laboratory data of PID patients. The ESID database consists of more than 200 disease specific registries, which are grouped into eight main categories. Each different PID has its own sub-registry. There is no national registry of PID in Turkey. A population prevalence of diagnosed PID in the United States at approximately 1 in 1,200 persons [14]. Data from the Kuwait National Primary Immunodeficiency Registry provides a PID prevalence estimate of 11.98 per 100 000 children and an estimated occurrence of PID is one in 1000 live births [8]. The Australian Society for Clinical Immunology and Allergy’s PID registry provided a prevalence of 4.9 per 100 000 in Australia and New Zealand combined [9]. The prevalance of PIDs in the general population has not been identified clearly in Turkey.
This study was performed to determine the frequency, characteristics and clinical course of various PID disorders among patients diagnosed over 6 years at the Department of Pediatric Immunology, Uludag University Medical Faculty and Department of Pediatric Immunology, Ege University Medical Faculty. These two centers are the major contributors reporting PID patients to ESID database from Turkey.
These data will help physicians to identify patients with PID and to provide a national database of PID in order to initiate a multi-institutional network study in Turkey. Additonally, we aimed to emphasize the importance of early diagnosis and treatment, to enhance the knowledge of PID among physicians, to determine the frequency of these diseases in Turkey.
Methods
Since 2004, patients are registered in ESID Database. The diagnosis was established according to the ESID criteria. Primary immunodeficiencies were classified according to the International Union of Immunological Societies’ (IUIS) criteria [4]. Uludag University Medical Faculty and Ege University Medical Faculty are the main referral centers for patients with PID, living in South Marmara Region and Aegean Region, respectively. These two universities are within the major participants of ESID Database from Turkey. The number of registered patients of these centes comprise almost 87.5 % of total registrations from Turkey (http://www.esid.org/statistics) [16]. The study protocol was approved by the Medical Ethics Committee of the Uludag University Medical Faculty and Ege University Medical Faculty.
Data collection was performed via the ESID Online Database for Primary Immunodeficiencies. Before entering patient data into the ESID Database informed consent was obtained from all participants and/or their guardians.
Laboratory analyses included complete blood count, peripheral blood smear, measurement of serum immunoglobulins (IgG, IgA, IgM, and IgE), isohemagglutinins, antibody response following immunization, delayed cutaneous hypersensitivity (purified protein derivative), peripheral blood lymphocyte subsets including T-cell subset (CD3, CD4, CD8), B-cell (CD19, CD20) and natural killer cell (CD56/16) by flow cytometry, IgG subclasses titer, lymphocyte proliferation test, neutrophil chemotaxis test, nitro blue tetrazolium dye test, and hemolytic titration of complement (CH50) as needed. Additionally, level of serum alpha fetoprotein, and assessment of the expression of CD18/CD11 on neutrophils by flowcytometry were performed. A deletion of the chromosome 22q11.2 region was determined by FISH. Both complement hemolytic activity (CH50) and most of the genetic testing were not available in our laboratories and hence we required support from research laboratories located abroad.
Statistical Analysis
Data were expressed as individual values or the mean ± SD for groups. Data analysis was performed using computer base and SPSS statistical software (version 17.0). A linear regression analysis was used to determine the association between birth date and diagnosis lag in months (duration from time of onset of disease to time of diagnosis).
Demographic statistics were obtained from Turkish Statistical Institute reports [17] The pointevalences were calculated using population data from January 2010.
Results
A total of 1435 patients (875 males, 560 females), including 1348 children and 87 adults, representing eight classes of PID were registered. We observed a similar gender distribution both among children and adults. Of these cases, 1360 patients were alive, while 75 were deceased or lost to follow-up. Among the 1360 living patients, 1275 were aged younger than 18 years and 85 patients were aged older than 18 years. In this study, patients were distributed in eight main categories of PID (Fig. 1).

Distribution of primary immunodeficiency disorders in Turkey
Overall, the onset of symptoms occurred at the mean age of 2,47 ±3,4 years (range49 months). The mean age at diagnosis was 4,71 ±5,1 years (range = 62 months). The mean duration from time of onset to time of diagnosis (diagnosis lag in months) in all groups was 26,9 ± 42,4 months (range 0.5–137 months). The age at onset, age at diagnosis, and diagnosis lag in months varied considerably for different types of PID (Table I). Of note, the combined T cell and B cell immunodeficiency diseases presented and were diagnosed at the youngest age and had the shortest diagnosis lag in months when compared to other groups.
There was an increasing trend towards the early recognition of PID in the past decade. Ninety six percent of all patients were diagnosed in the past 10 years (2000–2010) while 4 % were diagnosed before the year 2000. The diagnosis lag in months decreased from 83,6 months before the year of birth 1995 to 19,2 months since the year of birth 1995 (p < 0.001).
The estimated cumulative prevalence rate for all forms of PID was 30.5/100.000 in the general population. The prevalence rates are between 18.8/100.000 in Ege University and 45.2/100.000 in Uludag University. The major cause of this variation is most likely ascertainment bias because there may be differences in the criteria used in different centers for the diagnosis of some less severe forms of PID, such as transient hypogammaglobulinemia of infancy (THI). It was believed, however, that the majority of patients with antibody deficiency receiving IVIG were reported to the register. The prevalence rate for antibody deficiency is 22.7/100.000; the most common subtype is THI (7/100.000), followed by Ig A deficiency (IgAD) (5.6/100.000), Ig G subclass deficiency (4.6/100.000), common variable immunodeficiency (CVID) (1.39/100.000) and CSR defects and HIGM syndromes with unknown genetic cause (1.36/100.000). The prevalence rates of all immunodeficiency patients are given in Table II.
Consanguinity and Family History
One hundred eighty-eight patients (14.3 %) were products of consanguineous marriages, in most cases first cousins (Table III). Consanguinity was the most commonly seen in patients with immune dysregulation (80 %), combined T and B cell deficiency (54.5 %) and congenital defects of phagocytes (47.9 %). Of these patients, 17 were siblings from seven consanguineous families.
The first family who had two siblings with combined T and B cell deficiency, also had a previous sib death from infection. The second family had 5 cases with severe congenital neutropenia caused by Hax-1 gene defect. Two families had two children each of them with Wiskott Aldrich syndrome (WAS). Two families had two boys with XLA, one family also had two siblings death because of the same illness. Two brothers presented with CVID.
Clinical Features
PID patients showed a wide spectrum of clinical manifestations. Infectious complications, affecting 831 patients (58 %), were the most common, where the mean number of infection per year was 6.2. The most common subgroup suffering from infection was Combined T and B–cell immunodeficiencies (mean infection number per year: 20.1), followed by diseases of immune dysregulation and predominantly antibody deficiencies. Infection in all sites was the main presentation and acute lower respiratory infection was the most common infection, followed by sinusitis and otitis media. Disseminated BCG infection occurred in one case with SCID and four cases with IL12/IFNgamma pathway defects.
Autoimmune manifestations were seen in 32 of total (2.2 %), while most of them were CVID (17 patients), followed by WAS and severe congenital neutropenia. Vasculitis was the most common manifestation of autoimmunity, followed by autoimmune hemolytic anemia and autoimmune thrombocytopenia.
Twelve patients developed malignancy, including five non-Hodgkin lymphoma, where four out of five was Ataxia Telangiectasia and one X-linked lyphoproliferative disorder; two acute myeloid leukemia, where one patient had hyperimmunoglobulin M syndrome and the other had X-linked lyphoproliferative disorder; one acute lymphoblastic leukemia in CVID; one Hodgkin lymphoma in CVID; one Burkitt lymphoma in AID deficiency; and one hemangiopericytoma in WAS.
Genetic Analysis
Genetic analysis was performed in 116 patients (8 %). According to their diagnosis, genetic analysis was performed in 18 patients with combined immunodeficiency, 16 with chronic granulomatous disease, 13 with Di-George Syndrome, 9 with hyperimmunoglobulin E syndrome, 8 with severe congenital neutropenia, 8 with agammaglobulinemia, 8 with Mendelian susceptibility to mycobacterial disease, 7 with hyperimmunoglobulin M syndrome, 6 with Leukocyte Adhesion Deficiency, 5 with Wiskott Aldrich syndrome, 5 with osteopetrosis, 3 with FMF (Familial Mediterranean Fever), 2 patients with common variable immunodeficiency, 2 with chronic mucocutaneous candidiasis, 1 with APECED (autoimmune polyendocrinopathy) syndrome, 1 with Nijmegen Breakage Syndrome, 1 with Schimke Immunoosseus Dysplasia, 1 with Hyper IgD Syndrome, 1 with TRAPS, and 1 with Griscelli disease.
Treatment and Outcome of PID
209 (14.5 %) patients were on intravenous immunoglobulin replacement therapy. None of the patients received subcutaneous immunoglobulin because it is not available in our country. Forty-nine of antibody deficiency patients received oral antibiotic prophylaxis (76.6 %). Nineteen patients received bone marrow transplantation (seven SCID, four WAS, three Hyper Ig M syndrome, two osteopetrosis, two chronic granulomatous disease, one chronic mucocutaneous candidiasis) and sixteen of these patients were doing well. All ptients with phagocytic defects received antibiotic and anti-fungal prophylaxis and their survival rate was 100 %.
The mortality rate was 5.2 % which was mainly found in the group with combined T cell and B cell immunodeficiencies (SCID, 13 % of the total), and other well defined immunodeficiency syndromes (Ataxia Telangiectasia, 13 %.) The most common cause of death was sepsis (Table IV).
Discussion
Since 2004 ESID has been running a database for PID and more than 15.000 patients have been entered into the ESID Database until December 2011 [16]. Using the number of documented patients between 2006 and 2008, Gathmann et al. [15] reported that a minimum prevalence of PID in Turkey was 1.53 per 100.000 inhabitants. This number is relatively low due to the fact that documentation is far from complete. Many countries have their own registry. Previous reports of national and international gathering of information have brought epidemiological information and lots of important knowledge about immunodeficiencies.
In our study, although we have found the prevalence of total PID cases ascertained from the registery as 30.5/100.000 for two universities combined, the same data was found 45,2/100,000 in Uludag University and 18.8/100.000 in Ege University. First report from Kuwait National Primary Immunodeficiency registry showed that the prevalence of PID in children was 11.98/100.000 [8]. The estimated occurrence of PID in Turk ethnic group of northwestern Iran is about 24 per 100,000 live births [10]. This number also showed high frequency of PID in Turks.
National prevalence of all PID cases in Australia was 2.1/100.000 [5, 9]. French national registry centre (CEREDIH) showed a minimum prevalence of 4.4 patients per 100.000 inhabitants [18]. The overall prevalence of PIDs was found to range from 2.5 (Ireland) to 5.3 per 100,000 inhabitants (Norway) [11, 19]. The prevalence rate of our registry is much higher compare to those from the other countries. This is due to the fact that these two centers have registered all PID cases regularly and the consanguineous marriage rate in Turkey are more common compared to countries in Europe.
This study helps to our understanding of the prevalence and clinical presentation of primary immunodeficiencies in our region. Although 1435 patients were registered by us, documentation still lag behind in Turkey. Limited number of patients have been documented from many institutions. The common presentations of our PID patients were recurrent infections, especially in the respiratory tract. Both centers are specialized on pediatric cases, therefore fewer adult cases (% 6), most of which are congenital immunodeficiency disorders, are documented. Antibody deficiencies were seen in 73.9 % of the registered patients, followed by autoinflammatory disorders (13,3 %) and other well defined immunodeficiencies (5.5 %).
In this study, antibody deficiency was seen in two thirds of the patients, which is higher when compared to the patients reported from ESID database center in 2009 and reported from the Jeffrey Modell Foundation which give data on almost 80.000 PID patients from around the globe. [15, 20]. While transient haypogammaglobulinemia of infancy (THI) was the most common disease in antibody deficiencies in our series, CVID is the most prevalent disease of this group in ESID report and the PID registry from Ireland, Norway and Egypt [11, 13, 15, 19]. This discrepancy might be sourced that our centers specialized on the pediatric cases. More than two thirds of patients with CVID are diagnosed after second decade [21]. THI is a heterogeneous disorder characterized by reduced serum IgG (and sometimes IgA) level in the early childhood and may be associated with recurrent infections in some infants, although asymptomatic in others. A putative diagnosis was made after exclusion of other causes of hypogammaglobulinemia, while a definitive diagnosis of THI was made retrospectively in patients with normalized IgG levels and withdrawal of clinical symptoms that occurs usually between the 2nd and 4th years of life. The second common subtype of antibody deficiency in our study group was IgAD which is defined as very low levels of serum IgA (<0.05–0.07 g/L) in a patient older than 4 years of age with normal serum levels of IgG and IgM [21].
Both immunology centers namely Uludag University and Ege University also follow up rheumatologic patients. We have found 191 patients with Familial Mediterranean fever which is 13.3 % of total. Although FMF is an autosomal recessive disease, similar to the literature only in 12 % of our FMF cases were born to consanguineous parents. That is because FMF mutations are very common in the general population. FMF primarily affects populations originating in the Mediterranean region, particularly people of Armenian, Arabic, Turkish, and Jewish ancestry [22]. For these reasons we have found autoinflammatory disorders as a second largest group in our series.
In our study, the prevalence of combined T and B–cell immunodeficiencies was 0.58 in 100.000 The prevalence of predominantly T cell disorders was 1.4 in France and 1.6 in Israel [6, 18]. PID registry results from Iran which is a country sometimes has similar demographic features to our country showed the ratio of SCID 3.18 % in total, although they did not report the subtypes of SCID [7]. However, Shabestari et al. [10] reported that combined T and B–cell immunodeficiencies were the most common form of PID in Turk ethnic group of northwestern Iran. In contrast, the PID registry from South Africa, Egypt and ESID database center in the last report showed that T-B + was the most frequent disorder among SCID subtypes [13, 23]. T-B−SCID was found to be the most common disease in our group with combined T cell and B cell immunodeficiencies (2 % of total). Because of high frequency of consanguineous marriage in our country, we have rarely detected X-linked SCID which has been reported 75 in 227 of T-B + SCID patients in ESID database [15].
CGD was the most common phagocytic disorder which was supported by the reports from Iran, Thailand and ESID database [7, 15, 24]. Severe congenital neutropenia was the second common disorder in congenital defects of phagocytes. Registries from France and Norway reported that severe congenital neutropenia (SCN) is the most common one in phagocytic disorders [11, 18]. This discrepancy might be sourced that patients with SCN are also followed up by hematology centers beside immunology centers in our country.
The rate of consanguinity observed in our study is not much higher than the reported ones from Iran or Egypt or Kuwait [7, 8, 13]. There was a significant difference of consanguinity rate among the eight grups in our study. The consanguinity rate for diseases of immune dysregulation is 80 %, while in the predominantly antibody deficiencies subgroup the same is 9.7 % . In our study, 70 % of the patients are from this subgroup and therefore, the overall average of the consanguinity rate was found to be 14.3 %. Male predominance seen in our study is similar to registries from Iran, Kuwait, Israel and Australia [5–8].
Ig replacement represents one of the milestone therapeutic regimens in PID. The rate of Ig replacement was highest in the group of antibody-deficient patients, where 209 (14.5 %) of 1435 patients received this form of therapy. Hematopoietic stem cell transplantation (HSCT) is the only choice of radical treatment for children with a wide spectrum of primary immunodeficiencies (PIDs), but outcome is heavily dependent on the availability of a human leukocyte antigen-matched donor [1, 3]. In our series, 12 patients, mostly having SCID had undergone bone marrow transplantation. Eight of them are alive and still doing well.
The appropriate diagnosis and management would decrease health care cost and result in improved quality of life for these patients. Registries are being increasingly acknowledged as powerful tools for enhancing our knowledge of many features of disease (especially rare diseases), such as epidemiology, natural history, access to care and improvements in healthcare.
In our study, the delay in diagnosis was approximately 26,9 months. This number was 2.5 years in Kuwait [8]. However, our patients with combined T cell and B cell immunodeficiency were rapidly diagnosed in approximately five months. The reason for the shortest diagnosis lag was the early onset and severe symptoms of these patients in the first year of life. The overall mortality rate was 5.2 % which was mainly seen in the group with combined T cell and B cell immunodeficiencies and other well defined immunodeficiency syndromes.
Conclusion
Early diagnosis and treatment of PID are critical to minimizing morbidity and improving quality of life. The distributions of PID from several countries [9, 11, 13, 18, 20, 23], including Turkey, showed the highest prevalence in antibody deficiency diseases. In our study, the most common diseases in antibody deficiencies were THI and Ig A deficiency. The successful development of the registry in Turkey would help to estimate the burden of disease and should provide authorities with sufficient information to develop appropriate strategies to both diagnose and treat PIDs in Turkey.
References
Samarghitean C, Väliaho J, Vihinen M. IDR knowledge base for primary immunodeficiencies. Immunome Res. 2007;3:6.
Guzman D, Veit D, Knerr V, Kindle G, Gathmann B, Eades-Perner AM. The ESID Online Database network. Bioinformatics. 2007;23:654–55.
Marodi L, Notarangelo LD. Immunological and genetic bases of new primary immunodeficiencies. Nat Rev Immunol. 2007;7:851–61.
Geha RS, Notarangelo LD, Casanova JL, Chapel H, Conley ME, Fischer A. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776–94.
Baumgart KW, Britton WJ, Kemp A, French M, Roberton D. The spectrum of primary immunodeficiency disorders in Australia. J Allergy Clin Immunol. 1997;100:415–23.
Golan H, Dalal I, Garty B, Schlesinger M, Levy J, Handzel Z, et al. The incidence of primary immunodeficiency syndromes in Israel. IMAJ. 2002;4(Suppl):S868–71.
Rezaei N, Aghamohammadi A, Moin M, Pourpak Z, Movahedi M, Gharagozlou M, et al. Frequency and clinical manifestations of patients with primary immunodeficiency disorders in Iran: update from the Iranian primary immunodeficiency registry. J Clin Immunol. 2006;26:519–32.
Al-Herz W. Primary immunodeficiency disorders in Kuwait: First report from Kuwait national primary immunodeficiency registry. J Clin Immunol. 2008;28:186–93.
Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–24.
Shabestari MS, Maljaei SH, Baradaran R, Barzegar M, Hashemi F, Mesri A, et al. Distribution of primary immunodeficiency diseases in the Turk ethnic group, living in the northwestern Iran. J Clin Immunol. 2007;27:510–6.
Stray-Pedersen A, Abrahamsen T, Froland S. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000;20:477–85.
Matamoros Flori N, Mila Llambi J, Espanol Boren T, Raga Borja S, Fontan Casariego G. Primary immunodeficiency syndrome in Spain: First report of the national registry in children and adults. J Clin Immunol. 1997;17:333–9.
Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: A single-center study. J Clin Immunol. 2009;29:343–51.
Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497–502.
Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A. ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157(1):3–11.
The European Society for Immunodeficiencies: http://www.esid.org/statistics.php (2011).
Turkish Statistical Institute web site http://tuikapp.tuik.gov.tr/adnksdagitapp/adnks.zul
CEREDIH: The French PID study group. The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010;135:264–72.
Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: First report of the national registry in children and adults. J Clin Immunol. 2005;25:73–7.
Modell V, Gee B, Lewis DB, Orange JS, Roifman CM, Routes JM, et al. Global study of primary immunodeficiency diseases (PI)–diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol Res. 2011;51:61–70.
Report of a WHO scientific group. Primary immunodeficiency diseases. Clin Exp Immunol. 1997;109(1):1–28.
Savic S, Dickie LJ, Battellino M, McDermott MF. Familial Mediterranean fever and related periodic fever syndromes/autoinflammatory diseases. Curr Opin Rheumatol. 2012;24:103–12.
Naidoo R, Ungerer L, Cooper M, Pienaar S, Eley BS. Primary immunodeficiencies: A 27-year review at a tertiary paediatric hospital in Cape Town, South Africa. J Clin Immunol. 2011;31:99–105.
Benjasupattananan P, Simasathein T, Vichyanond P, Leungwedchakarn V, Visitsunthorn N, Pacharn P, et al. Clinical characteristics and outcomes of primary immunodeficiencies in Thai children: an 18-year experience from a tertiary care center. J Clin Immunol. 2009;29:357–64.
Acknowledgements
We are thankful to Benjamin Gathmann for his help on providing access to our ESID registry data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kilic, S.S., Ozel, M., Hafizoglu, D. et al. The Prevalances and Patient Characteristics of Primary Immunodeficiency Diseases in Turkey—Two Centers Study. J Clin Immunol 33, 74–83 (2013). https://doi.org/10.1007/s10875-012-9763-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-012-9763-3