
Abstract
Chronic granulomatous disease (CGD) results from an inherited defect in the phagocytic cells of the immune system. It is a genetically heterogenous disease caused by defects in one of the five major subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. There is a paucity of data from India on CGD. We herein describe the clinical features in 17 children with CGD from a single tertiary referral center in India. A detailed analysis of the clinical features, laboratory investigations and outcome of 17 children 7 with X-linked (XL) and 10 with autosomal recessive (AR) form was performed. Diagnosis of CGD was based on an abnormal granulocyte oxidative burst evaluated by either Nitroblue Tetrazolium (NBT) test or flow cytometry based Dihyrorhodamine 123 assay or both. The molecular diagnosis was confirmed by genetic mutation analysis in 13 cases. The mean age at diagnosis and the age at onset of symptoms was significantly lower in children diagnosed with XL- CGD compared those with AR disease. Mutations were detected in CYBB gene in 6 patients with XL-CGD and NCF-1 gene mutations were observed in 7 cases of AR- CGD. The course and outcome of the disease was much worse in children diagnosed with X-linked form of disease compared to AR forms of the disease; 4/7 (57 %) children with X-CGD were dead at the time of data analysis. This is one of the largest series on chronic granulomatous disease from any developing country.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic granulomatous disease (CGD) is an inherited and genetically heterogenous immunodeficiency disorder resulting from defects of one of the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex in phagocytic cells. It is a rare disease affecting between 1in 2,00,000 and 1 in 2,50,000 live births [1]. The actual incidence is likely to be higher due to underdiagnosis of patients presenting with milder disease phenotype. CGD was initially described in 1954 [2] and 1957 [3], but it was not well characterized as a distinct clinical entity until 1959 [4].
NADPH oxidase complex is composed of five major subunits. Two of these gp91phox (cytochrome b-245 β polypeptide) and p22phox (cytochrome b-245 α polypeptide) are membrane bound components encoded by the CYBB gene and the CYBA gene respectively. The remaining three components of the complex include p47phox, p67phox and p40phox encoded by the corresponding genes namely, NCF1 (neutrophil cytosolic factor 1), NCF2 (neutrophil cytosolic factor 2) and NCF4 (neutrophil cytosolic factor 4) [5]. All the components of the NADPH oxidase except for the gp91phox are not phagocyte specific and expressed in other tissues as well [6]. Therefore defects in the components other than gp91phox may have subtle effects on other tissues.
The NADPH oxidase complex catalyzes the conversion of molecular oxygen O2 to superoxide anion (O2 -) and other reactive oxygen intermediates. Therefore, defects in any of the components of the NADPH oxidase complex results in impaired killing of intracellular microorganisms and renders patients with CGD susceptible to recurrent and often life threatening infections with bacteria and fungi. X-linked recessive form of the disease due to mutations in the CYBB gene encoding for gp91phox accounts for approximately 65 % of patients with CGD. Mutations in the NCF1 gene encoding for the p47phox account for 30 % of the cases whereas CYBA and NCF2 mutations are detected in <5 % patients each. Only one patient with mutation in NCF4 has been reported thus far [7]. The risk of mortality in CGD is estimated to be 1–5 % annually and is likely dependent on the mode of inheritance i.e. X-linked or AR.
There is a paucity of data on chronic granulomatous disease from developing countries although large series have been published from Europe and USA. The clinical spectrum of disease including the type of infections, frequency of breakthrough infections, morbidity and mortality are likely to be different in the context of a developing country. Hence we embarked to perform a retrospective analysis of our cohort of chronic granulomatous disease diagnosed and managed over the last 2 decades.
Patients and methods
A detailed data analysis of 17 children diagnosed with chronic granulomatous disease from August 1993 to April 2013 was performed. The study was conducted in the Pediatric Allergy and Immunology, Unit, Advanced Pediatrics Centre and the Department of Immunopatholgy, Postgraduate Institute of Medical education and Research (PGIMER), Chandigarh. Our Institute serves as tertiary level referral centre for North West India . The study was approved by the Department Review Board in consonance with the existing practice at our institution. Data were retrieved from the case records and files of the Pediatric Immunodeficiency Clinic at the Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research (PGIMER). Evaluation of the clinical manifestations included the age at presentation and diagnosis, presenting complaints and detailed laboratory parameters.
Diagnosis of CGD was based on an abnormal granulocyte oxidative burst evaluated by either Nitroblue tetrazolium test [8, 9] (NBT, n = 10) or flow cytometry based Dihydrorhodmaine (DHR)123 assay [10, 11] (n = 1) or both (n = 6). These tests were also performed on the parents and siblings where available (n = 9) to determine the mode of inheritance. The NBT test was performed using leucocyte rich plasma whereas heparin anticoagulated whole blood was used for the DHR assay . Phorbol Myristate acetate (PMA) was used for stimulation of neutrophils in the DHR assay and yeast cells and/or PMA where used for stimulation in the NBT dye reduction test.
Complete blood count including a total and differential leucocyte count, mean platelet volume, absolute lymphocyte count, eosinophil and neutrophil counts were determined in each case using a five part automated hematology analyzer.
Serum Immunoglobulins IgG, A and M were estimated by end-point nephelometry using a semi-automated nephelometer MININeph (The Binding Site, Birmingham, UK). Lymphocyte subset analysis was done using BD Tritest™ CD45PerCP, CD3-FITC and CD19- PE antibody cocktail from BD Biosciences (San Jose, USA) following a lyse no wash protocol. Briefly 50 μl of EDTA anticoagulated blood was added to 20 μl of the antibody cocktail in a FACS tube, mixed, vortexed and solution. After 10 min the tubes were vortexed and acquisition was performed on a BD FACSCalibur flow cytometer.
Investigations also included X-rays and CT scan when indicated.
Genetic mutation analysis results was performed in 13 cases. The mutation analysis was not done in four cases because some of the children were diagnosed when facilities for mutation studies were not available or the children had died before these studies could be performed. The mutation analysis was conducted at the National Defense Medical College, Saitama, Japan and at the Department of Pediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong after obtaining written informed consent from the parents.
Results
Seventeen patients (15 males and 2 females) from 15 families with a diagnosis of CGD were included in the analysis. 7 children (41 %) children were diagnosed with X-linked form of CGD and 10 (59 %) were found to have an AR form of disease (Table I). Definite mutation analysis was available in 13/17 patients. Mutations were detected in CYBB gene was detected in 6 patients classified as definite X-CGD. Similarly mutations in NCF-1 gene were detected in 8 patients (definite AR-CGD). One case had a history of loss of 3 elder male siblings and this was classified as presumed X-CGD. Two cases with no mutation in CYBB, NCF-1 and CYBA genes were presumed to be AR-CGD cases. The mean age for onset of symptoms was 1 year 8 months (median 10 months, range 1 month - 8 years) and a diagnosis of CGD was made at a mean age of 4 years 6 months (median 3.0 years, range 7 months -16 years). The mean age at diagnosis was significantly lower in children diagnosed with X-linked CGD compared those with autosomal recessive disease: mean 3 years 8 1/2 months (median 1 year, range 7 months–16 years) versus mean 5 years (median 3 years 6 months, range 1 year–10 years) respectively. Similarly the age at onset of symptoms was also different in two groups mean 2 years ½ months (median 3 months, range 1 month–12 years) versus mean 1 year 10 months (median 1 year 3 months, range 1 1/2 months–5 years) (Table II).
Infectious Complications
All patients in the present cohort received prophylactic antimicrobials in the form of co-trimoxazole and Itraconazole or Ketoconazole after diagnosis. None of the patients received therapy with recombinant IFN-γ, because of financial constraints. Infectious episodes were managed with appropriate antimicrobials and antifungal agents including the newer antifungal agents such as voriconazole often for prolonged periods.
Pneumonia and recurrent lymphadenitis were the commonest clinical manifestations, present in 82.3 % (14/17) (Figure 1 and Table III). Recurrent skin and/or subcutaneous abscesses were found in 47 % (8/17) children. Hepatomegaly was detected in 59 % (10/17). In addition, liver abscesses were observed in 23.5 % (4/17) of children, in the absence of significant hepatomegaly in two children. Gastrointestinal symptoms in the form of loose stools, abdominal pain and distention were present in 35 % (6/17). Septicemia was detected in 25 % (4/16) children, albeit pre-terminally in 3 children. Bone and joint involvement was detected in 3 children. Perianal/ischiorectal abscess was also seen in 2 children. Other less common manifestations included recurrent otitis media and renal involvement, found in two children and one child respectively . Recurrent ulcerative stomatitis was also observed in one child (Table III).

Predominant clinical manifestations in children with CGD
Microorganims Isolated From Patients
An infectious etiology could not be established in most infectious episodes. Culture reports were often negative despite repeated blood and urine culture, culture of bronchoalveolar lavage whenever required on clinical grounds, fine needle aspirates and biopsy specimens probably because most of these children had received antimicrobials, including antifungals in some cases, prior to culture. Polymerase chain reaction based tests were not performed for etiological diagnosis because of lack of availability of the same. Aspergillus species was the most frequently isolated microorganism, being isolated from lung, synovium and blood in 6 patients. Apart from the common species i.e. A fumigatus, A flavus, some rarer species such as A terreus were also isolated. Candida species were also isolated in a significant proportion of cases (3/17). Fungal serology for Aspergillus was positive in four children (Table IV).
Staphyloccocus aureus was isolated from culture of pus in Patient 6 who preented with recurrent and multiple liver abscesses. Burkholderia cepacia was isolated from blood culture in one child with fulminant septicemia (Patient 2) and in one case Mucor was identified along with Mycobacterium tuberculosis from a resected segment of lung (Figure 2) (Patient 5). In addition Fusarium dimerum was cultured from the sputum in a child with autosomal recessive form of disease (Patient 16).

a Photomicrograph showing epithelioid granulomas with multinucleate giant cells. b Photomicrograph depicting angioinvasion (H&E) ×40
Non Infectious Complications
Two X-CGD patients presented with abdominal pain and distention without any evidence of an infectious etiology. Patient 8 presented with features of intestinal obstruction with abdominal distention and pain. This was initially attributed to sepsis, the antimicrobials were changed twice. However when he did not respond to this regimen, steroids were given to which he responded dramatically with improvement in the gastrointestinal symptoms. However subsequently he developed respiratory distress and succumbed to his illness.
Patient 1 with X-linked form of the disease presented with fever, hepatosplenomegaly and persistent anemia, thrombocytopenia, hyperferritinemia (1,013 ng/ml) and mild hypertriglyceridemia (301 mg/dl) on follow-up. Bone marrow examination showed evidence of hemophagocytosis. His fibrinogen level was however normal at 2.4 g/l. A diagnosis of hemophagocytic lymphohistiocytosis (HLH) was made based on the clinical features and laboratory parameters. He responded well to immunosuppressive therapy in combination with intravenous vancomycin, meripenem and voriconazole.
Patient 4 presented with renal involvement in the form of mildly deranged renal function tests and hydronephrotic changes in the right kidney.
Investigations
Most of the children had panhypergammaglobulinemia with mean IgG level of 1571.5 ± 450 g/dl, mean IgM level of 232.9 ±101.2 g/dl and a mean IgA level of 252.8 ± 179 g/dl. Lymphocyte subsets were within normal range in most children.
Nitroblue Dye Reduction Test [8, 9]
Leucocyte rich plasma was separated from heparin anticoagulated blood from both test and control subjects. Two drops (100 μl) each of leucocyte rich plasma was placed on 2 different glass slides labeled as “stimulated” and “unstimulated” respectively. Two drops of (100 μl) of a 0.2 % Nitroblue tetrazolium dye was added to each slide. One drop of a 20 % suspension of Baker’s yeast was added to the slide labeled as “stimulated”. Both the slides were then placed in a humified chamber at 37 °C for 30 min. The slides were taken out after 30 min, cover slips were placed on both them and they were examined under a light microscope.
Neutrophils showing blue black cytoplasmic clumps of formazan were counted in both stimulated and unstimulated tests. At least 100 neutrophils were counted and the results were expressed as a percentage of cells showing the blue black granules. The procedure was repeated similarly with the control sample. Normally 85–95 % of stimulated neutrophils showed reduction of nitroblue tetrazolium to blue black formazan. Tests in which less than 30 % of stimulated neutrophils showed no reduction were interpreted as abnormally low and suggestive of chronic granulomatous disease. In some cases 2 μl of (100 μg/ml) of Phorbol myristate acetate was used to stimulate the neutrophils.
Dihydrorhodamine Assay [10, 11]
Staining Procedure
Four tubes, two each for normal control and patient sample were labeled as unstimulated and stimulated. Hundred (100 μl) of heparin anticoagulated blood was added to each tube. 1 μl of 1 mM Dihydrorhodamine (Sigma Cat No D1054) in DMSO was added to each tube and incubated for 15 min at 370C in a water bath. 2 μl of Phorbol-12- myristate-13- acetate (100 μg/ml) was added to the tubes labeled stimulated for both test and control samples. The tubes were further incubated for further 15 min at 370C and then lysed using BD FACS lysing solution for 10 min. The tubes were then centrifuged at 1,500 rpm for 5 min. Wash the cells with Phosphate buffered saline and spun again at 1,500 rpm for 5 min and decant supernatant. This procedure was repeated twice.
Acquisition and Data Analysis
Samples were acquired on FACS Calibur or FACS CANTO flow cytometer. Threshold was adjusted to exclude debris and neutrophils were gated by Forward Scatter(FSC)/Side scatter (SSC) gating. At least 10,000 gated events were acquired and recorded for each tube. Mean fluorescence intensities (MFIs) of stimulated and unstimulated samples and fold change in MFIs for both test and control samples was recorded.
Interpretation
The assay was performed in 7/17 patients. Four (4) of these patients were AR-CGD cases with a mutation in NCF-1 gene. Two of these were presumed AR-CGD (Patient 7 and 9) and one patient was a case of X-CGD with a mutation in the CYBB gene. The mean of mean fluorescence intensities in the unstimulated state in test was 52.6 ± 59.2 and in the stimulated state was 68.9 ± 78 and mean fold change was 1.23 ± 0.2. In the control the mean MFI in the unstimulated tube was 39.82 ± 52 and in the stimulated tube was 627. 16 ± 880 and the mean fold change was 21.75 ± 18.1 A diagnosis of CGD was considered when there was no shift in the histograms after stimulation with PMA and minimal or no change in MFIs in unstimulated vs stimulated states (Table V) (Fig. 3a–d).

Histograms from a DHR assay showing MFIs in gated neutrophils (a) Unstimulated patient (b) Post stimulation with PMA patient (c) Unstimulated normal control (d) Post stimulation with PMA normal control
We could not estimate resdiual NADPH oxidase activity as superoxide production was not estimated in any of the cases.
Molecular Basis of the Defects
Molecular defects could be identified in 13 of the 17 patients with CGD. The mutation analysis could not done in 4 cases because some of the children were diagnosed when facilities for mutation studies were not available or the children had died before these studies could be performed. A mutation in the CYBB gene was detected in 6 children with X-linked CGD in which the mutation analysis was performed. All the six mutations detected in the CYBB gene were point mutations. Four of these six point mutations were nonsense mutations and 2 were missense mutations. The two missense mutations in the CYBB gene were novel and both mutations were deleterious using the SIFT program for the prediction of protein change. Same type of mutation in NCF1 gene encoding for p47phox protein (c. 73_74delGT, p.Tyr26HisfsX25) were detected in 7 of the 8 children with AR- CGD in whom mutation analysis was performed (Table I).
Mortality and Cause of Death
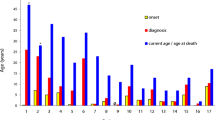
Six of the 17 patients (35 %) had succumbed at the time of the analysis. Four of these were boys who had X-linked CGD (Patients 2, 3, 5, and 8) and Patient 15 who was a sister of patient 14 with AR-CGD with mutation in NCF1 gene had also died. Patient 7 with presumed AR-CGD had also succumbed to his illness at time of analysis. Median age at death was 1 year 5 months with a range of 7 months–9 years. The median age at death in children with XL-CGD was 10 months with a range of 7 months–2 years. Three children with X-linked CGD died of fulminant septicemia. Another child (Patient 5) with X-linked CGD died of a massive pulmonary bleed following decortication to remove a segment of the lung for Mucormycosis. One girl (Patient 15) with autosomal recessive CGD and another child with presumed AR-CGD (Patient 7) died due to fulminant pneumonia at the age of 14 years and 2 years respectively. Mean survival in X-CGD was estimated to be 31.23 months (95 % CI: 5.1, 57.8 months) while the mean survival in AR-CGD was 176.6 months (95 % CI : 72.2, 281 months). This difference in survival was found to be close to level of statistical significance by Log Rank (Mantel-Cox) test (p = 0.065) (Fig. 4).

Survival and follow-up in AR-CGD vs X-CGD
Discussion
Chronic granulomatous disease has been infrequently been reported from India with only occasional case reports [12–15]. However in these case reports a presumptive diagnosis of CGD was made on the basis of NBT dye reduction test without a confirmed diagnosis by genetic analysis. The present study is to best of our knowledge the largest case series of CGD from India with a well-characterized molecular defect in 13 of the 17 patients. Thirteen cases of CGD have been reported from another tertiary care centre in North India in a period of 2 years between July 2004–August 2006 [16]. However all the cases in this study where diagnosed on the basis of NBT dye reduction test, DHR assay was not performed and the diagnosis was not confirmed by a genetic analysis for the putative genes in CGD in any of these patients.
AR-CGD was more common in the present series detected in 58 % of children compared to X-linked form of disease which was detected in 42 % of children. This is in contrast to previous studies from Europe and USA in which X-CGD. [1, 17, 18] and similar to a large series reported from Turkey [19] and another series from Tunisia [20]. Consanguinity is not frequent in North India as has been reported from the series reported from Turkey and Tunisia. Consanguinity was found in 2/17 patients in our cohort and both had AR-CGD with mutation in the NCF-1 gene. However the pattern of marriages is largely endogamous, with marriages being restricted within closed knit communities and this may result in a higher incidence of different autosomal recessive diseases in different population groups [21]. The mean age at diagnosis was 4 years 6 months whereas the mean age of onset of symptoms was 1 year 8 months and children with X-linked CGD were diagnosed earlier than AR- CGD, similar to cohorts reported earlier[22, 23]. Clinical manifestations were also similar to what has been reported earlier with rare and interesting findings in two cases [22–24].
Hemophagocytic lymphohistiocytosis (HLH) is a rare and potentially fatal complication of CGD. Only few cases of HLH complicating the course in CGD have been reported thus far. Apart from an increased susceptibility to recurrent infections by intracellular microorganisms, CGD is also associated with hyperinflammation and proinflammatory cytokine milieu, which could predispose these children to HLH. Most cases of HLH in children with CGD are secondary to infections mainly with Burkholderia cepacia and Leishmania. An isolated case with a perforin gene variant has also been reported [25–27].
One of the children had pulmonary mucormycosis (Fig. 2a and b) along with a co-infection with Mycobacterium tuberculosis. This child presented with a mass in the right hemithorax eroding the ribs and extending into the mediastinum. Mucormycosis has rarely been reported in patients with CGD, almost exclusively in those receiving prolonged and significant immunosuppression [26, 27]. However this child had not received any form of immunosuppressive therapy, highlighting that mucormycosis can occur in CGD even without immunosuppression.
Mycobacterial infections both due to BCG and M. tuberculosis have been reported in patients with CGD from China, Iran and Latin America [28, 29]. Mycobacterium was detected in only one of our patients, diagnosed by detection of acid fast bacilli in a lung biopsy specimen. Although BCG vaccine is administered at birth in India only one child in our cohort developed suppuration at site of BCG administration followed by swelling at the root of the neck. The apparently low incidence of BCG and Mycobacterium tuberculosis infection in our cohort of CGD patients could be due to under detection. Moreover, some of the children presenting pneumonic consolidation did receive antitubercular therapy based on a presumptive diagnosis of tuberculosis. Two children in our cohort were diagnosed as tuberculosis based on the finding of granulomatous inflammation in lung biopsy specimens despite no isolation of Mycobacterium on culture or demonstration of acid-fast bacilli in the biopsy specimens. Recently germline mutations in the CYBB gene that selectively impair the activity of the NADPH oxidase complex in the monocyte-derived macrophages but not monocytes and neutrophils and predisposition to infection by Mycobacteria tuberculosis have been reported [30].
The spectrum of microorganisms isolated in the present series were similar to those reported in earlier studies [24, 31, 32]. Aspergillus was the most common organism isolated in our cohort followed by Staphylococcus aureus, Burkholderia cepacia and Candida. Several previous studies have shown that Aspergillus is emerging as the single most significant pathogen for infectious complications and mortality in patients with CGD. In North America, majority of infections in CGD are caused by five microorganisms namely Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia and Aspergillus species [1].
All the presently known mutations in the CYBB gene have been reported in a large series recently [33]. It comprised of 1,267 unrelated kindreds with 1,415 patients. Six hundred and eighty one different mutations were identified in the patients and 487 (73.1 %) of these mutations were unique for one kindred. Single nucleotide substitutions were the commonest mutations reported. The missense mutations, splice site mutations and nonsense mutations were observed in 21.3 %, 17.6 % and 14.1 % respectively. Deletions contributed to 35.6 % of all mutations and insertions to 8 % of the mutations. All the mutations in CYBB gene in our cohort were single nucleotide substitutions; 66.7 % were nonsense mutations and 33.3 % were missense mutations. Two novel deleterious missense mutations were detected in our cohort.
Similarly a large series of all known mutations in the genes implicated in AR-CGD have also been published [34]. All the patients with AR- CGD in our cohort were found to have a same mutation in the NCF1 gene encoding for p47phox protein although they were unrelated. Mutation in NCF1 gene is the commonest cause of AR- CGD and accounts for approximately 20–25 % of all cases. All the patients also had the same mutation in the NCF1 gene i.e. GT dinucleotide deletion in exon 2. This particular mutation has been reported in more than 60 patients worldwide with AR-CGD due to mutation in NCF1 gene and 97 % of the alleles [35]. There are 2 pseudo NCF1 genes with a GT deletion in each of them. The preponderance of this mutation in patients can be explained by multiple recombination events between the functional NCF1 gene and these closely linked pseudogenes, each gene having multiple recombination hot spots such as Alu repeats, Chi sequence and human mini-satellite repeats [36].
The overall mortality (35 %) was higher than what has been reported previously. The mortality in a large cohort of 429 patients from Europe was 20 % [23] whereas in an Italian cohort the mortality was 13 % [22]. This high mortality could be attributed to several factors. None of our patients received recombinant interferon γ. All patients were managed on cotrimoxazole and itraconazole prophylaxis along with management of breakthrough infections. The possibility of increased exposure to infectious agents in a developing, tropical country might have also contributed to a higher incidence of infections.
A higher frequency as well as greater severity of breakthrough infections compared to developed countries could have contributed to the increased mortality. Delay in initiating therapy for intercurrent infections due to the parents having to travel long distances to reach medical facilities and poor economic conditions are likely to have contributed significantly to this higher mortality. Haemotopoietic stem cell transplantation was not performed in any of our patients.
Finally although the total number of patients in the present cohort is very small to draw definite conclusions, it was observed that the course and outcome of the disease was much worse in children diagnosed with XL-CGD compared to AR forms of the disease. Four of the seven children (4/7) with X-CGD were dead at the time of data analysis. Three of these four children had nonsense mutations in the CYBB gene resulting in a stop codon as has been reported previously [1, 23, 29]. However it has also been conclusively shown that residual NADPH oxidase activity determined largely by the specific mutation in any of genes responsible for CGD rather than the gene itself is a useful predictor of outcome and survival [37]. It can also be concluded from this cohort that it is possible to provide a reasonable quality of life to patients with prophylactic antimicrobials even in a developing country like ours with all its constraints.
References
Winkelstein JA, Marino MC, Johnston Jr RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79(3):155–69.
Janeway CACJ, Davidson M, Downey W, Gitlin D, Sullivan JC. Hypergammaglobulinemia associated with severe, recurrent, and chronic non-specific infection. Am J Dis Child. 1954;88:388–92.
Berendes H, Bridges RA, Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med. 1957;40(5):309–12.
Bridges RA, Berendes H, Good RA. A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome. Am J Dis Child. 1959;97(4):387–408.
Kuijpers T, Lutter R. Inflammation and repeated infections in CGD: two sides of a coin. Cell Mol Life Sci. Jan;69(1):7–15.
Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;2006(349):re8.
Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114(15):3309–15.
Park BH, Holmes BM, Rodey GE, Good RA. Nitroblue-tetrazolium test in children with fatal granulomatous disease and newborn infants. Lancet. 1969;1(7586):157.
Baehner RL, Nathan DG. Quantitative nitroblue tetrazolium test in chronic granulomatous disease. N Engl J Med. 1968;278(18):971–6.
Emmendorffer A, Hecht M, Lohmann-Matthes ML, Roesler J. A fast and easy method to determine the production of reactive oxygen intermediates by human and murine phagocytes using dihydrorhodamine 123. J Immunol Methods. 1990;131(2):269–75.
Roesler J, Hecht M, Freihorst J, Lohmann-Matthes ML, Emmendorffer A. Diagnosis of chronic granulomatous disease and of its mode of inheritance by dihydrorhodamine 123 and flow microcytofluorometry. Eur J Pediatr. 1991;150(3):161–5.
Salaria M, Singh S, Kumar L, Datta U, Sehgal S. Chronic granulomatous disease. Indian Pediatr. 1999;36(6):594–6.
Nair PS, Moorthy PK, Suprakasan S, Jayapalan S, Preethi K. Chronic granulomatous disease. Indian J Dermatol Venereol Leprol. 2005;71(3):199–201.
Pinto LM, Udwadia ZF. A 24-year-old man with giddiness, hemoptysis, and skin lesions. Chest. 2008;134(5):1084–7.
Soneja M, Batra A, Vikram NK, Ahuja A, Mohan A, Sood R. Actinomycosis and nocardiosis co-infection in chronic granulomatous disease. J Assoc Physicians India. Apr;60:66–8.
Verma S, Sharma PK, Sivanandan S, Rana N, Saini S, Lodha R, et al. Spectrum of primary immune deficiency at a tertiary care hospital. Indian J Pediatr. 2008;75(2):143–8.
Ahlin A, De Boer M, Roos D, Leusen J, Smith CI, Sundin U, et al. Prevalence, genetics and clinical presentation of chronic granulomatous disease in Sweden. Acta Paediatr. 1995;84(12):1386–94.
Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr. 2000;137(5):687–93.
Koker MY, Camcioglu Y, van Leeuwen K, Kilic SS, Barlan I, Yilmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. Jul 30.
El Kares R, Barbouche MR, Elloumi-Zghal H, Bejaoui M, Chemli J, Mellouli F, et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J Hum Genet. 2006;51(10):887–95.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461(7263):489–94.
Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 2008;126(2):155–64.
van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234.
Finn A, Hadzic N, Morgan G, Strobel S, Levinsky RJ. Prognosis of chronic granulomatous disease. Arch Dis Child. 1990;65(9):942–5.
van Montfrans JM, Rudd E, van de Corput L, Henter JI, Nikkels P, Wulffraat N, et al. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr Blood Cancer. 2009;52(4):527–9.
Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer. Mar;56(3):460–2.
Alvarez-Cardona A, Rodriguez-Lozano AL, Blancas-Galicia L, Rivas-Larrauri FE, Yamazaki-Nakashimada MA. Intravenous immunoglobulin treatment for macrophage activation syndrome complicating chronic granulomatous disease. J Clin Immunol. Apr;32(2):207–11.
Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27(3):224–30.
Bustamante J, Aksu G, Vogt G, de Beaucoudrey L, Genel F, Chapgier A, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol. 2007;120(1):32–8.
Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. Mar;12(3):213–21.
Hasui M. Chronic granulomatous disease in Japan: incidence and natural history. The Study Group of Phagocyte Disorders of Japan. Pediatr Int. 1999;41(5):589–93.
Mouy R, Fischer A, Vilmer E, Seger R, Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114(4 Pt 1):555–60.
Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. Oct 15;45(3):246–65.
Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. Apr 15;44(4):291–9.
Casimir CM, Bu-Ghanim HN, Rodaway AR, Bentley DL, Rowe P, Segal AW. Autosomal recessive chronic granulomatous disease caused by deletion at a dinucleotide repeat. Proc Natl Acad Sci U S A. 1991;88(7):2753–7.
Gorlach A, Lee PL, Roesler J, Hopkins PJ, Christensen B, Green ED, et al. A p47-phox pseudogene carries the most common mutation causing p47-phox- deficient chronic granulomatous disease. J Clin Invest. 1997;100(8):1907–18.
Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. Dec 30;363(27):2600–10.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rawat, A., Singh, S., Suri, D. et al. Chronic Granulomatous Disease: Two Decades of Experience From a Tertiary Care Centre in North West India. J Clin Immunol 34, 58–67 (2014). https://doi.org/10.1007/s10875-013-9963-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-013-9963-5