
Abstract
Solution NMR studies of α-helical membrane proteins are often complicated by severe spectral crowding. In addition, hydrophobic environments like detergent micelles, isotropic bicelles or nanodiscs lead to considerably reduced molecular tumbling rates which translates into line-broadening and low sensitivity. Both difficulties can be addressed by selective isotope labeling methods. In this publication, we propose a combinatorial protocol that utilizes four different classes of labeled amino acids, in which the three backbone heteronuclei (amide nitrogen, α-carbon and carbonyl carbon) are enriched in 15N or 13C isotopes individually as well as simultaneously. This results in eight different combinations of dipeptides giving rise to cross peaks in 1H–15N correlated spectra. Their differentiation is achieved by recording a series of HN-detected 2D triple-resonance spectra. The utility of this new scheme is demonstrated with a homodimeric 142-residue membrane protein in DHPC micelles. Restricting the number of selectively labeled samples to three allowed the identification of the amino-acid type for 77 % and provided sequential information for 47 % of its residues. This enabled us to complete the backbone resonance assignment of the uniformly labeled protein merely with the help of a 3D HNCA spectrum, which can be collected with reasonable sensitivity even for relatively large, non-deuterated proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Twenty-five years after the introduction of 1H–13C–15N triple-resonance pulse sequences in solution NMR (Ikura et al. 1990a; Kay et al. 1990b; Montelione and Wagner 1990) methods for the backbone resonance assignment of uniformly isotopically labeled proteins have become well-established. As a consequence of an inherently low chemical shift dispersion in combination with large linewidths, protocols routinely used for soluble proteins are, however, less straightforward when applied to α-helical membrane proteins (Kim et al. 2009; Maslennikov and Choe 2013; Nietlispach and Gautier 2011; Sanders and Sönnichsen 2006; Tamm and Liang 2006). In particular, crucial triple-resonance experiments that involve 13C–13C magnetization transfer steps such as HNCACB (Wittekind and Mueller 1993) or HN(CA)CO (Clubb et al. 1992) often suffer from low sensitivity. Also, amino-acid type selective experiments (Dötsch et al. 1996; Feuerstein et al. 2012; Lescop et al. 2008; Pantoja-Uceda and Santoro 2008; Rios et al. 1996; Schubert et al. 1999) that would lead to a considerable spectral simplification, employ pulse sequences comprising more coherence transfer steps or editing elements and are therefore often unsuitable for larger systems. In a different approach, selective 15N labeling provides amino-acid type information (Griffey et al. 1985; Klammt et al. 2004; LeMaster and Richards 1985; Muchmore et al. 1989; Ozawa et al. 2004, 2006) while dual selective labeling with 15N and 13C isotopes additionally permits an identification of specific amino acid pairs via the interresidue 15N i –\( ^{13} {\text{C}}_{i - 1}^{\prime } \) scalar coupling (Griffey et al. 1986; Ikura et al. 1990b; Kainosho and Tsuji 1982; Rule et al. 1993; Shi et al. 2004; Tate et al. 1992; Weigelt et al. 2002; Yabuki et al. 1998). NMR methods that rely on amino-acid specific isotope labeling usually feature decreased susceptibility to the adverse effects of slow molecular tumbling and conformational exchange.
Use of cell-free expression systems allows a cost-efficient production of selectively labeled protein samples and to a large extent avoids problems due to isotope scrambling (Klammt et al. 2004; Michel et al. 2013; Ozawa et al. 2004; Shi et al. 2004; Sobhanifar et al. 2010; Staunton et al. 2006; Yabuki et al. 1998). Nevertheless, combinatorial methods (Ozawa et al. 2006; Parker et al. 2004; Shortle 1994; Sobhanifar et al. 2010; Staunton et al. 2006; Wu et al. 2006) are required to keep the number of samples to be prepared at a minimum, while gaining comprehensive site-specific information to achieve sequential backbone assignments with a high degree of confidence. Combinatorial dual-selective labeling with 15N and 1-13C enriched amino acids which involves recording of 15N-HSQC and 2D HN(CO) spectra has been successfully applied to the assignment of membrane proteins (Maslennikov et al. 2010; Trbovic et al. 2005). Recently introduced combinatorial triple-selective labeling (Löhr et al. 2012) augments such labeling schemes with doubly 13C/15N enriched amino acids resulting in an increased amount of residue types as well as sequential pairs that can be identified with a given number of samples. The new combinations containing 13C/15N amino acids either in position i or i − 1 of a dipeptide or in both can be deconvoluted by the presence/absence of 1H–15N i cross peaks in additional 2D triple-resonance spectra such as HN(CA), HN(COCA), (CO)HN(CA), and DQ-HN(CA).
Here we present a refined combinatorial protocol which employs four different classes of labeled amino acids including exclusively at the 2-(Cα) position 13C-labeled ones. This results in a significantly increased percentage of amide groups for which sequential information is available, thus serving as anchor points during backbone assignment. Since the signature of 15N-labeled residues preceded by 2-13C-labeled residues in the set of 2D triple-resonance spectra acquired in the context of combinatorial triple-selective labeling is identical to that of 13C/15N-labeled residues preceded by unlabeled ones, a supplementary experiment is required to distinguish the two cases. The latter relies on the one-bond 13Cα–\( ^{13} {\text{C}}^{\prime } \) scalar coupling to filter out signals originating from the fully 13C/15N-labeled species. In the application shown here virtually complete backbone assignments could be obtained using a single complementary 3D HNCA spectrum, recorded on a uniformly labeled sample, to fill the gaps between the selectively labeled residues.
Materials and methods
Sample preparations
Production of selectively and uniformly labelled samples of TMD0 was carried out using a continuous-exchange cell-free expression system based on an E. coli S30 extract (Schwarz et al. 2007). The protein, consisting of 142 amino acid residues and a C-terminal His10-tag, was cloned into a pIVex2.3-MCS vector. Optimization of the construct for cell-free expression, protein purification and screening of sample conditions for NMR have been described previously (Tumulka et al. 2013). Per NMR sample, 2.3 ml reaction mixture were dialyzed against 34.5 ml feeding mixture yielding about 2 mg of purified protein. Amino-acid type compositions of selectively labeled samples are summarized in Table 1. The remaining amino acid types were supplied in unlabeled form. The amino acid concentration in the cell-free reaction was 0.5 mM for the reaction mixture and 0.6 mM for the feeding mixture, corresponding to total amounts of 6–14 mg of labeled amino acids per sample. For expression of the uniformly labeled sample, 64 mg of a mix containing all 20 amino acids fully 13C and 15N labeled was employed. Final protein concentrations varied between 0.5 and 0.6 mM (monomer concentrations) in 190 μl sodium acetate buffer (pH 5), containing 100 mM NaCl and 1 % dihexanoyl-phosphatidylcholine (diC6PC). Samples were placed in 4-mm susceptibility-matched Shigemi tubes that were inserted into standard 5-mm tubes, filled with a 0.8-mM solution of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) in D2O.
NMR spectroscopy
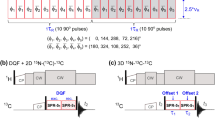
All pulse sequences employed in the current study were of the [15N, 1H]-TROSY (Pervushin et al. 1997; Salzmann et al. 1998) type and included sensitivity-enhanced gradient echo/antiecho coherence selection (Czisch and Boelens 1998; Kay et al. 1992; Pervushin et al. 1998; Weigelt 1998). Bandselective 1H pulses centered in the amide region were used to accelerate longitudinal relaxation between scans according to the BEST protocol (Lescop et al. 2010; Schanda et al. 2006). In order to constructively add polarization from proton magnetization longitudinally relaxed during the pulse sequence in the subsequent scan, acquisition periods were followed by a 15N 180° pulse (Favier and Brutscher 2011). As previously described (Löhr et al. 2012) 2D HN(CO), HN(CA), HN(COCA), and DQ-HN(CA) pulse sequences were derived from the corresponding three-dimensional schemes (Bax and Ikura 1991; Bayrhuber and Riek 2011; Ikura et al. 1990a; Nietlispach et al. 2002) by replacing 13C evolution periods by fixed 3-μs delays and, where applicable, omitting 1H 180° decoupling pulses. In 3D HNCA (Ikura et al. 1990a) and HNCACB (Wittekind and Mueller 1993) experiments, proton decoupling during 13C evolution was achieved by a pair of 1H broadband inversion pulses (Smith et al. 2001) applied at t 1/4 and 3t 1/4. A novel carbonyl-filtered (CO-filtered) 2D HN(CA) pulse scheme, shown in Fig. 1, was applied to distinguish correlations involving 13C/15N- and 2-13C-labeled amino acids. The sequence includes a 13Cα spin-echo period with a duration of 1/(2 1 J(\( ^{13} {\text{C}}^{\prime } \),13Cα) in which the latter coupling can evolve or not, depending on the position of a \( ^{13} {\text{C}}^{\prime } \) 180° pulse (see figure legend for details). The two subspectra were recorded in an interleaved manner and contain either all HN(CA) correlations if the coupling is not active (control spectrum), or a subset containing only those involving 2-13C-labeled residues if the coupling is active (filtered spectrum).

Carbonyl-filtered 2D BEST-[15N, 1H]-TROSY-HN(CA) pulse sequence for distinction of \( ^{13} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \) and \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \) moieties in 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \)-selectively labeled proteins. Radiofrequency (RF) pulses with flip angles of 90° and 180° are represented by filled and open symbols, respectively. All proton and nitrogen pulses are centered in the amide region (ca. 8.5 and 119 ppm, respectively). The 13C carrier frequency is placed in the Cα region, its exact position being determined by the 13Cα chemical shift distribution of the 2-13C and 13C/15N-labeled amino acid types of the respective sample to which the sequence is applied. The widths of shaped 1H and 13C pulses given in the following are valid for 800 MHz 1H Larmor frequency and must by scaled by 800/X for applications at X MHz spectrometers. The initial two proton 90° pulses have a PC9 shape (Kupče and Freeman 1994) and a width of 2.2 ms, while the third and fourth use 1.4-ms time-reversed E-BURP-2 and regular E-BURP-2 shapes (Geen and Freeman 1991), respectively. A RE-BURP (Geen and Freeman 1991) shape and a width of 1.5 ms is employed for 1H 180° pulses. Rectangular pulses on α-carbons are applied with an RF field of ν/151/2 for 90° flip angle and ν/31/2 for 180° flip angle, where ν is the difference between 13Cα and 13C’ offsets in Hz (Kay et al. 1990b). The 180° 13Cα pulse in the center of period ε has an R-SNOB shape (Kupče et al. 1995) and duration of 0.6 ms. Carbonyl-selective 90° and 180° pulses of 0.1 ms duration are applied at 176.5 ppm using phase modulation (Boyd and Soffe 1989; Patt 1992) and have an amplitude envelope corresponding to the center lobe of a sin(x)/x function. Carbonyl-filtered and control spectra in which a 180° \( ^{13} {\text{C}}^{\prime } \) pulse is applied at positions A and B, respectively, are recorded alternately. The carbonyl 180° pulse labeled with an asterisk compensates for Bloch–Siegert phase shifts induced by the former. Pulses on 15N are applied at the highest available power level. Phases are cycled according to ϕ1 = 2(y), 2(−y); ϕ2 = x, −x; ϕ3 = 4(x), 4(−x); ϕ4 = 8(x), 8(−x); ϕrec = x, 2(−x), x, −x, 2(x), 2(−x), 2(x), −x, x, 2(−x), x. Delay durations are adjusted as follows: τ = 5 ms, Δ = 24 ms, ε = 9.4 ms, τG = 0.38 ms. The t 1 evolution time is implemented in a semi-constant time manner (Grzesiek and Bax 1993; Logan et al. 1992), where χ = Δ/t 1,max. Pulsed field gradients have a sine-bell shape and are applied along the z-axis using the following durations and peak amplitudes: G1, 0.3 ms, −10 % (percentage of the maximum available gradient strength, i.e. ca. 70 G/cm); G2, 0.3 ms, −15 %; G3, 0.3 ms, −12 %; G4, 1.4 ms, 78.9 %; G5, 0.3 ms, 8 %; G6, 1.0 ms, 25 %; G7, 0.175 ms, 64 %. For each t 1 increment N- and P-type transients are collected alternately by inverting the polarity of G4 and changing pulse phases ϕ5 from y to −y and ϕ6 from x to −x. The two FIDs are stored separately and then added and subtracted to form the real and imaginary parts of a complex data point with a 90° zero-order phase shift being added to one of the components (Kay et al. 1992). Phase ϕ4 is inverted along with the receiver reference phase in every other increment to shift axial peaks to the edge of the spectrum in the 15N dimension. The Bruker pulse sequence code and acquisition parameters for an 800 MHz spectrometer are provided in the Supplementary Material
Experiments were performed at a sample temperature of 323 K on Bruker Avance spectrometers equipped with cryogenic 1H{13C/15N} triple-resonance probes. For selectively labeled samples, BEST-TROSY type HSQC, HN(CA), DQ-HN(CA) and CO-filtered HN(CA) spectra were acquired at 800 MHz, HN(CO) and HN(COCA) spectra at 600 MHz, and 3D HNCA and HNCACB spectra on [U-13C; 15N]-labeled protein at 950 MHz. The interval between the end of acquisition and the beginning of the next scan was 0.5 s in HSQCs and 0.3 s in all 2D and 3D triple-resonance experiments. Spectral widths were 11 and 50 ppm in the 1H and 15N dimensions, respectively, of all 2D experiments. Here, the 1H acquisition time was adjusted to 43.6 ms, while 15N acquisition times were 63.2 ms (256 complex t 1 increments) in HSQC, 52.6 ms (160 increments) in HN(CO), 43.5 ms (176 increments) in HN(CA), 42.1 ms (128 increments) in HN(COCA), 24.7 ms (100 increments) in DQ-HN(CA), and 31.6 ms (128 increments) in CO-filtered HN(CA) experiments. The number of transients accumulated per FID in experiments on the three combinatorial selectively labeled samples were 16, 64, 96, 176, and 512 in HSQC, HN(CO), HN(CA), HN(CO)CA, and DQ-HN(CA), respectively, resulting in measurement times of 1 h, 2h45′, 4h25′, 6h5′, and 13h40′. Carbonyl-filtered HN(CA) spectra were acquired with different number of scans (and corresponding measurement times) for individual samples, i.e. 416 (28h30′) for sample 1, 256 (17h30′) for sample 2, and 512 (35 h) for sample 3, where the number of scans applies to filtered and control subspectra each and the measurement time is the total duration of both experiments. Differing number of scans/measurement times were employed in HN(CO)CA (256/8 h), DQ-HN(CA) (1520/41 h), and CO-filtered HN(CA) (320/23 h) experiments on the additional (“test”) sample, containing only four labeled amino-acid types (Table 1).
Acquisition times in 3D HNCA and HNCACB experiments were 43.6 ms (1H), 44.9 ms (15N), and 8.3/7.0 ms (13C, HNCA/HNCACB). Both experiments employed a non-uniform sampling scheme in which 22.8 % of the data points of the full t 1/t 2 grid were acquired. Measurement times were 46 h (HNCA) and 91 h (HNCACB) using 64 and 72 scans/FID, respectively.
Spectra processing and analysis was performed with TopSpin 3.2 (Bruker). Cosine-squared window functions were applied for apodization in all dimensions. Spectra were referenced with respect to internal DSS using consensus Ξ values for 13C and 15N (Wishart et al. 1995). Contour levels were drawn on an exponential scale using a factor of 21/2 for preparation of all plots shown in the following.
Results and discussion
Motivation for the development of an improved selective labeling scheme
Traditionally, the backbone resonance assignment of [13C; 15N]-labeled proteins relies on matching intraresidual and sequential cross peaks in three-dimensional experiments that correlate amide 1H and 15N chemical shifts with those of 13Cα (Ikura et al. 1990a), \( ^{13} {\text{C}}^{\prime } \) (Clubb et al. 1992), or 13Cβ (Wittekind and Mueller 1993) nuclei. With increasing molecular size, however, these experiments become less efficient due to rapid transverse relaxation, and at the same time spectral overlap becomes more severe unless the intrinsic chemical shift dispersion of the protein is favorable. A combination of both difficulties is usually encountered for α-helical membrane proteins. Replacement of protons with deuterons significantly decelerates dipolar relaxation of adjacent nuclei and, to some extent also reduces overlap owing to line narrowing. On the downside, expression of deuterated proteins requires additional optimization, is usually accompanied by lower yields and is comparatively expensive. Furthermore, problems can occur due to incomplete back-exchange of amide protons, partial protonation at Cα positions and reduced protein stability. As an alternative, a method termed combinatorial triple-selective labeling that exclusively relies on information obtained with non-deuterated protein samples has been introduced (Löhr et al. 2012). The method was validated with the voltage sensor domain of the voltage-dependent K+ channel from Aeropyrum pernix (KvAP), a 149-residue protein for which resonance assignments have previously been obtained using perdeuterated samples (Butterwick and MacKinnon 2010; Shenkarev et al. 2010), and was additionally employed to overcome problems due to signal overlap in 3D spectra recorded on uniformly labeled samples of proteorhodopsin. Here we propose a refined combinatorial selective labeling protocol and apply it for the de novo assignment of the N-terminal transmembrane domain of the lysosomal peptide transporter TAPL, named TMD0.
TMD0 is not essential for the homodimerization of the ABC half-transporter TAPL nor for peptide transport activity of the transporter. However, it harbors the lysosomal targeting motif and represents the interaction site with the lysosomal associated membrane proteins LAMP-1 and LAMP-2 (Demirel et al. 2010, 2012). TMD0 is predicted to consist of four helices and exists as a homodimer in diC6PC micelles, which tumbles with a rotational correlation time of 25 ns at a temperature of 50 °C as determined using a two-dimensional version of the [15N, 1H]-TRACT experiment (Lee et al. 2006). To assess the feasibility of obtaining backbone assignments without deuteration, 3D HNCA and HNCACB experiments were performed on a [U-13C; 15N]-labeled protein sample. Projections onto the 1H-15N planes of both spectra are presented in Fig. 2a, b. The HNCA is of relatively high quality, showing correlations for all amide groups that are also detectable in a simple 2D HSQC. By contrast, the HNCACB, albeit recorded in about twice the measurement time, is practically useless, since only a few cross peaks from mobile residues are observed. A 3D HN(CA)CO experiment was not attempted here because its sensitivity is usually similar to that of an HNCACB.

Application of three-dimensional triple-resonance experiments to [U-13C; 15N]-labeled TMD0. Spectra were recorded at 950 MHz using non-uniform sampling. Projections along the 13C dimensions of 3D HNCA and 3D HNCACB spectra are shown in a, b, respectively. The HNCACB experiment was performed with a 13Cα–13Cβ transfer time of 11 ms, optimized to observe 13Cβ resonances. c, d 1H (F 3)–13C (F 1) strips from the 3D HNCA with a width of 0.2 ppm along the 1H dimension, taken at the 15N (F 2) chemical shifts given at the top. Assignments of intra- and interresidual cross peaks, resulting from analysis of the current spectrum in conjunction with combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \)-selective labeling, are indicated. Dashed lines trace the sequential walk in d
Considering the generally poor signal dispersion of α-helical membrane proteins and the limited amino-acid type information available from 13Cα chemical shifts, the question arises whether unambiguous sequential connectivities can be established solely on the basis of a 3D HNCA. This problem is illustrated in the lower part of Fig. 2, showing HNCA correlations of well-resolved backbone amides of TMD0. All 1H-13C strips in Fig. 2c contain intraresidual cross peaks involving lowfield 13Cα resonances (between approximately 67 and 68 ppm), indicative for either valine or threonine residues, and interresidual cross peaks with chemical shifts ranging from 58.0 to 64.7 ppm, leaving the amino acid type of the sequentially preceding residue undetermined. Clearly, analysis of the 3D HNCA would benefit from prior identification of residue types or, preferentially sequential pairs. Application of the combinatorial labeling method described below revealed that two of the seven amide groups in Fig. 2c belong to threonines, the remaining ones representing valines. For V16, V26, and V89 the amino acid types of the preceding residues were determined as well, allowing for only two or three possible assignments in each case. No additional information was obtained for V93 and V99, except that certain preceding residue types could be ruled out. For instance, unlike V89, V99 is found not to be preceded by a leucine although the two HNCA strips show correlations at almost identical 13Cα chemical shifts. Owing to the unique occurrence of the respective pairs, the two threonines were sequence specifically assigned following identification of the preceding residue types. The second example (Fig. 2d) encompasses a continuous stretch of ten residues near the C-terminus of TMD0, all of which have 13Cα chemical shifts in the amino-acid type unspecific 58–64 ppm range. The spectral quality of the 3D HNCA for most of these amides is only moderate, with rather low intensities of sequential peaks. In addition intraresidual and sequential cross peaks for residues F131, L133, W135, and L137 partially overlap. Taken together, it would be quite challenging to find the correct sequential connectivities without prior knowledge of at least individual residue types. However, with the exception of S138, combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling identified the amino acid types of the entire stretch, localized the two leucine–leucine pairs, and provided three anchor points (F131, W135, and T139) for sequential assignment. With this information at hand, evaluation of the 3D HNCA became comparatively straightforward.
Merits of combinatorial selective labeling
Amino acid specific isotope labeling considerably simplifies NMR spectra and is a powerful tool to aid the resonance assignment process of proteins (Butterwick and MacKinnon 2010; Griffey et al. 1985; Hagn and Wagner 2014; LeMaster and Richards 1985; McIntosh and Dahlquist 1990; Michel et al. 2013; Ozawa et al. 2004; Reckel et al. 2008; Takeuchi et al. 2007; Vajpai et al. 2008a). For instance, single-selective 15N labeling readily allows the identification of amino acid types even for large systems as it only requires recording of sensitive HSQC spectra. On the other hand, obtaining exhaustive amino acid type information entails preparation of up to 19 NMR samples, if the protein of interest contains all non-proline amino acid types. Sequence-specific amide group assignments are directly accessible via dual-selective labeling with 15N and carbonyl (1-13C) labeled amino acids, pioneered by Kainosho and Tsuji (1982) which has been demonstrated to be extremely useful for monitoring individual sites by measuring HNCO-type spectra (Gossert et al. 2011; Griffey et al. 1986; Rule et al. 1993; Tate et al. 1992; Weigelt et al. 2002; Yabuki et al. 1998). Aiming at full backbone assignments, separate samples with different pairs of amino acids would have to be prepared, and data to be collected on, to an extent of the same order of magnitude as the number of residues present in the protein, which is clearly unfeasible. To this end, recently reviewed combinatorial methods (Jaipuria et al. 2012) using multiple mixtures, each containing different subsets of labeled amino acids rather than a single of each labeling type, are much more efficient (Parker et al. 2004; Shi et al. 2004; Shortle 1994; Staunton et al. 2006; Trbovic et al. 2005; Wu et al. 2006).
Although the production of selectively labeled protein samples by cell-free expression is comparatively rapid and inexpensive it is desirable to minimize the number of NMR samples in a combinatorial scheme. This can be achieved by increasing the amount of information to be retrieved from each sample. Including fully 13C/15N-labeled amino acids in addition to 15N- and 1-13C-labeled ones allows the identification of more amino acid types and more sequential pairs, because they can be observed themselves in 1H–15N correlation spectra (position i in a given dipeptide) and as the preceding residue (position i − 1) of a 15N-labeled amino acid (Löhr et al. 2012). Compared to dual-selective combinatorial labeling (Maslennikov et al. 2010; Trbovic et al. 2005) the number of detectable isotopomeric dipeptide species is raised from two (\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i and \( ^{13} {\text{C}}_{i - 1}^{\prime } \)–15N i ) to six (\( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \), \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{13} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \), \( ^{13} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{13} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \), \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \), \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{13} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \), \( ^{13} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{13} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \)). To take advantage of this larger variety it is necessary to distinguish between 15N and 13C/15N-labeled residues in position i and between 1-13C and 13C/15N-labeled residues in position i − 1. In principle, the former distinction can be made on the basis of signal intensities when different enrichment levels of the two species are employed (Parker et al. 2004). However, taking into account the pronounced dynamic range often observed in spectra of membrane proteins, this method may not always be fully reliable in applications to the latter. Alternatively, the individual species can be selected spectroscopically by means of 2D HN(CA) (to distinguish 15N i from 13C/15N i residues) and 2D HN(COCA) (to distinguish 1-13C i−1 from 13C/15N i−1 residues) experiments, recorded in addition to 15N-HSQC and 2D HN(CO) spectra. A fifth experiment, DQ-HN(CA) exclusively detects signals from sequential pairs of the same, 13C/15N-labeled amino acid type, which cannot be spotted via dual-selective 15N, 1-13C labeling.
Concept of combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling
The labeling scheme described here expands on the combinatorial triple-selective labeling method (Löhr et al. 2012) that involves acquisition of two-dimensional BEST-TROSY type (Farjon et al. 2009; Favier and Brutscher 2011) HSQC, HN(CO), HN(CA), HN(COCA), DQ-HN(CA) and, optionally, (CO)HN(CA) spectra on three samples, each containing four 15N-labeled, one 13C/15N-labeled, and two 1-13C-labeled amino acid types. It allows identification of ten amino acid types along with deconvolution of six different combinations of labeling types in a dipeptide fragment. In an attempt to enhance the amount of sequential information without increasing the number of samples we now extend the labeling pattern by including two amino acid types per sample that are selectively 13C-labeled at the α-carbon (2-position), resulting in the scheme listed in Table 1. In such samples, 15N-labeled amino acids preceded by 13C/15N-labeled ones give rise to cross peaks in each of HN(CO), HN(CA), and HN(COCA) spectra, whereas those preceded by 1-13C- and 2-13C-labeled amino acids are exclusively observed in HN(CO) and HN(CA) spectra, respectively. One complication, however, arises because 13C/15N-labeled residues preceded by non-13C-labeled ones are likewise detected in HN(CA) experiments only. In order to distinguish \( ^{13} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \) from \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \) moieties, a new 2D experiment, depicted in Fig. 1, must be performed in addition to those mentioned above.
The HN(CA)-type pulse sequence of Fig. 1 contains a 13Cα spin-echo period, adjusted to 1/(2 1 J(\( ^{13} {\text{C}}^{\prime } \),13Cα) following the 15N–13Cα magnetization transfer. When the second \( ^{13} {\text{C}}^{\prime } \) 180° pulse is applied at position A (carbonyl-filtered version) transverse 13Cα coherence is converted into an antiphase term in the presence of an adjacent \( ^{13} {\text{C}}^{\prime } \) spin and will not be converted into observable magnetization during the remainder of the sequence. In addition it is canceled by cycling of pulse phase ϕ3 along with the receiver. Consequently, no intraresidual signal will be obtained for 13C/15N-labeled amino acids. In the case of an interresidual 15N–13Cα magnetization transfer between 15N- and 2-13C-labeled amino acids, however, 13Cα coherence “survives” the filter element due to the absence of a \( ^{13} {\text{C}}^{\prime } \) coupling partner, thus giving rise to a cross peak in the CO-filtered HN(CA) spectrum. It must be mentioned that, as a result of transverse relaxation during the additional spin-echo period of ≈9 ms duration, the sensitivity of the CO-filtered HN(CA) experiment is significantly lower than that of the conventional HN(CA), especially for slowly tumbling proteins. Therefore, it cannot be ruled out that a cross peak due to a 2-13C i−1/15N i pair is missed in the CO-filtered HN(CA) although it was observed in the conventional HN(CA). If the number of signals detected in the CO-filtered HN(CA) is lower than that expected based on the amino acid sequence, it is advisable to record a control experiment with identical parameters. In this control experiment, the \( ^{13} {\text{C}}^{\prime } \) 180° pulse in the sequence of Fig. 1 is applied at position B, preventing evolution of the 1 J(\( ^{13} {\text{C}}^{\prime } \),13Cα) coupling. The absence of a cross peak in the control spectrum hints at the possibility that its absence in the filtered version is caused by relaxation rather than by the action of the filter, thus avoiding misinterpretation due to false negatives. A special situation is met when a 13C/15N-labeled amino acid is preceded by a 2-13C-labeled one. In principle, such pairs are also observable in the CO-filtered HN(CA), but their intensity is strongly attenuated by the passive 1 J(15N,13Cα) coupling competing with the usually smaller 2 J(15N,13Cα) coupling during the magnetization transfer to the 2-13C-labeled residue. Uniquely yielding cross peaks in DQ-HN(CA) but not in HN(CO) or HN(COCA) spectra, 2-13C i−1/\( ^{15} {\text{N}}_{i}^{13} {\text{C}}_{i}^{\alpha } \) combinations are however readily identified. The correlation between all possible pairs of labeled and unlabeled amino acids and the occurrence of cross peaks in the series of 2D spectra is summarized in Table 2.
NMR samples containing individual 15N or 13C/15N residue types together with amino acids selectively 13C labeled at the 1- and/or 2-positions have previously been utilized to complement assignment information obtained from uniformly labeled protein (Butterwick and MacKinnon 2010; Vajpai et al. 2008b), but the simultaneous incorporation of all four classes in a combinatorial manner to our knowledge has not been reported so far. The approach applied here may be viewed as a “two-dimensional” combinatorial method, where in the “sample dimension” residue types containing a 15N label are identified from the pattern of presence/absence of cross peaks in HSQC spectra of each sample, and in the “spectroscopic dimension” the various combinations of 12C/13C isotopomeric dipeptide species are edited via the presence/absence of cross peaks in a series of 2D HN(C)-type triple-resonance spectra. In this regard there is a loose analogy to an assignment strategy that combines a precursor-based selective unlabeling protocol with Hadamard-encoded amino acid-type editing to enhance the information that can be gained from a limited set of samples (Rasia et al. 2012).
Application to TMD0
Labeling schemes such as the one shown in Table 1 are to a large extent protein specific. The choice of individual amino acid types requires optimization with respect to the attainable information which in turn depends on a protein’s primary structure. Obviously, of highest importance is the selection of the subset of residues that are either 15N or 13C/15N labeled, as for all of them at least the residue type will be determined. In the application describe here we opted mostly for methyl-containing and aromatic residue types that have a high occurrence in the hydrophobic, structured regions of membrane proteins, and were found to be largely undetectable in the 3D HNCACB spectrum and to have a low dispersion of 13Cα chemical shifts in the 3D HNCA of TMD0. In situations where the resolution enhancement of highly congested spectral regions is pivotal, as for instance in denatured proteins or IDPs, it might be more rewarding to include less abundant amino acid types, especially in the case of the 15N-labeled one that is present in all samples (Wu et al. 2006). Preferably, the three 13C/15N labeled species should cover (i−1, i)-pairs of identical amino acid types, if present in the sequence, because these would not be identified with any other combination of labels. In a second step, the two sets of three amino acid types that are either 1-13C or 2-13C labeled must be defined. To this end, it is inevitable to scan the amino acid sequence on a pair-by-pair basis to find those combinations that maximize the number of detectable dipeptides. Using the permutations of two out of three selectively 13C-labeled amino acids types in each of the three samples (cf. Table 1), unambiguous 1-13C i−1–15N i and 2-13C i−1–15N i pairs are obtained for those 15N-labeled amino acids that are present in two or in all three samples. In general, this is not the case for 15N-labeled amino acids present in one sample only, as well as for the three 13C/15N-labeled amino acids in position i. However, not all of the possible combinations may in fact occur in a given amino acid sequence. A judicious choice of the 1-13C and 2-13C labeled amino acid types and their distribution among the three samples should therefore take into account the missing combinations in order to lift the twofold ambiguities as much as possible. Finally, combinations that would potentially be unambiguously identified, but are missing in the sequence, should be avoided when designing a combinatorial labeling scheme.
Before presenting spectra of TMD0 utilized for 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling aided backbone assignment, two features of the protein must be mentioned. First, not uncommon for membrane proteins, there is a large dynamic range in HSQC signal intensities between residues in mobile loop regions and residues located in well-structured regions or affected by conformational-exchange induced line broadening. The effect is amplified in triple-resonance spectra due to longer periods in the pulse sequences, during which the underlying differential relaxation takes place. As a consequence, cross peaks of 15N labeled amino acids producing the most intense signals in HSQCs are sometimes observed unexpectedly in triple-resonance spectra at 13C natural abundance. Their intensities can be of similar magnitude as the weakest “true” correlations. i.e. those expected on the basis of the respective 13C labeling pattern. While the appearance of natural abundance signals can potentially lead to some confusion, they are usually readily sorted out precisely because of their high intensity in HSQC spectra. Second, despite extensive optimization of sample conditions (Tumulka et al. 2013) several residues give rise to a second, minor amide signal. The majority of these amino acids are located in loop regions in the vicinity of proline residues, implying that cis–trans isomerism of the latter might be responsible for the occurrence of two sets of signals. In each case, the assignment of the minor peaks could be verified by their presence in the same set of 2D triple-resonance spectra as the corresponding major peaks in combination with near-identical 13Cα chemical shifts in the 3D HNCA.
To assess the viability of the modified labeling scheme, a “test” sample containing only one amino acid type per class was prepared, resulting in easily manageable spectra as shown in Fig. 3. The [15N-Lys; 13C/15N-Val; 1-13C-Tyr; 2-13C-Ala]-labeled sample of TMD0 comprises all dipeptide moieties listed in Table 2. The amino acid sequence includes four lysine and 15 valine residues, hence, a total of 19 cross peaks are expected in the HSQC. All correlations are indeed observed (Fig. 3a), with three residues (V7, V8, and V107) giving rise to duplicate peaks, as mentioned above. As a first step in 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \)-labeling based assignment, signals from 15N and 13C/15N-labeled amino acid types need to be distinguished. Signals of all 13C/15N-labeled amino acids are expected in the HN(CA), whereas 15N-labeled amino acids are only detected if they are preceded by either 13C/15N or—as a new feature—2-13C-labeled residues. In accordance with the amino acid sequence of TMD0, the HN(CA) of the test sample (Fig. 3b) contains correlations from 17 residues, i.e. those of the 15 valines, as well as one Val–Lys and one Ala–Lys pair. The Val–Lys pair is differentiated from valines preceded by unlabeled amino acids by the presence of cross peaks in HN(CO) (Fig. 3c) and HN(COCA) (Fig. 3d) spectra while the Ala–Lys pair is identified with the help of the novel CO-filtered HN(CA) experiment (Fig. 3e). The two Val–Val pairs in TMD0 (V7–V8 and V8–V9) are readily identified because, in contrast to the Val–Lys combination, they are detectable in the DQ-HN(CA) spectrum (Fig. 3f). The additional cross peaks in the DQ-HN(CA) (V7 and V20) indicate Ala–Val combinations because only 13C/15N-labeled residues preceded by 2-13C-labeled ones are observed in DQ-HN(CA) and are absent in HN(CO)/HN(COCA) spectra (cf. Table 2). Note that a long accumulation time (41 h) was required to detect residues V7 and V8 in the DQ-HN(CA) because both are split into two separate peaks.

Identification of the various dipeptide isotopomers in a 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeled protein sample. Shown are HSQC (a), HN(CA) (b), HN(CO) (c), HN(COCA) (d), CO-filtered HN(CA) (e), and DQ-HN(CA) (f) spectra of a 0.5-mM sample of diC6PC-solubilized [15N-Lys; 13C/15N-Val; 1-13C-Tyr; 2-13C-Ala]-labeled TMD0 recorded at 800 (a, b, e, f) or 600 MHz (c, d). Sequence-specific assignments of the four lysine residues follows from the presence/absence of cross peaks (positions indicated by colored circles) in the individual spectra, as explained in the text. The corresponding dipeptide labeling patterns are shown beneath (g), where 15N and 13C nuclei are highlighted in blue and red, respectively. Asterisks mark signals from a small fraction of unfolded protein present in the sample
Each of the four 15N-labeled lysine residues in TMD0 is part of a different dipeptide labeling pattern in the test sample (Fig. 3g), as reflected by the presence or absence of cross peaks in the spectra of Fig. 3a–f. In detail, K2 (position highlighted by yellow circles) is sequentially preceded by an unlabeled residue and therefore exclusively detected in the HSQC. Being preceded by a 1-13C-labeled residue, K5 (orange circles) is identified by an additional cross peak in the HN(CO) spectrum. Correlations due to K100 (circled in magenta) are observed in HSQC, HN(CA), HN(CO), and HN(COCA) spectra, which is the fingerprint of a 15N-labeled residue preceded by a 13C/15N-labeled one. Finally, the presence of an HN(CA) cross peak in the absence of HN(CO) and HN(COCA) cross peaks (green circles) either points at an isolated 13C/15N-labeled residue or at a 15N-labeled residue preceded by a 2-13C-labeled one. The detection of a signal in the CO-filtered HN(CA) reveals that the latter option applies in the case of K70. Note that K5 and K100 would have been identified already with the previous triple-selective labeling scheme (Löhr et al. 2012), but it was only after introducing the additional 2-13C label that an unequivocal sequence-specific assignment of all lysine residues in TMD0 was achieved.
Spectroscopic distinction of the eight isotopomeric dipeptide types (Table 2) in a set of three 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeled samples (Table 1) is performed in a fully analogous manner as described above for the test sample. However, owing to its combinatorial nature (Jaipuria et al. 2012), such a labeling scheme does not simply multiply the information content of the latter by the number of samples. Rather, it permits including four 15N-labeled and two of each 1-13C- and 2-13C-labeled amino acid types per sample. In the following, the utility of combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \)-selective labeling to establish anchor points for sequential NH assignment of TMD0 is demonstrated.
Two well-resolved regions of the entire grid of 2D proton–nitrogen correlation spectra obtained for the three samples are shown in Fig. 4. Following identification of the 13C/15N-labeled amino acid type in each sample (Ile, Trp, and Leu) from the pattern observed in triple-resonance spectra, the amino acid type of 15N-labeled residues is determined by inspection of the three HSQC spectra. In the region of Fig. 4a, five signals are observed in each of the HSQCs and, hence, must be due to threonine residues (cf. Table 1). Signals arising from phenylalanine and arginine are identified by their appearance in the HSQCs of samples 1 and 2 and samples 1 and 3, respectively. Cross peaks due to valine residues are detected in the spectra of sample 3 exclusively. In the final step, the signal occurrence in the triple-resonance spectra is employed to derive information about the respective sequentially preceding residue type. In the region of Fig. 4a, T87 and T123 are observed in HN(CO), HN(COCA), and HN(CA) spectra of samples 1 and 2, respectively, and hence are preceded by a 13C/15N-labeled residue. The corresponding Ile–Thr and Trp–Thr pairs are unique in the sequence of TMD0 and, therefore, unambiguously assigned. The unique Ser–Thr pair is identified by the exclusive observation of HN(CO) peaks of T139 in samples 2 and 3. Threonine residues 10 and 65 give rise to correlations in both regular and CO-filtered HN(CA), but not in HN(CO) and HN(COCA) spectra, indicating they are preceded by 2-13C-labeled residues. Being present in the spectra of samples 1 and 2 (T10) and 1 and 3 (T65) the latter residue types are identified as valine and alanine, respectively. While Ala–Thr is a unique pair, Val–Thr occurs twice in the sequence, nonetheless providing valuable information for subsequent analysis of a 3D HNCA. The sole arginine signal in this region (R57) arises from a residue that is preceded by a 2-13C-labeled residue type in the current labeling scheme. The fact that the expected HN(CA) peaks are observed in the spectra of sample 3 and not in those of sample 1 or both points at tyrosine as the preceding residue type, allowing its distinction from the two Val–Arg pairs in the TMD0 sequence and therefore its immediate sequential assignment. Two of the 13C/15N-labeled isoleucine residues of sample 1 are preceded by 2-13C-labeled ones and are therefore in principle detectable in the CO-filtered HN(CA) spectrum. Whether they are actually observed in the latter is a matter of sensitivity and, as explained above, depends on transverse 15N relaxation times as well as one-bond and two-bond 15N–13Cα couplings constants. In the present case, a cross peak due to I86 is in fact observed, whereas the one due to I24 is not. However, since cross peaks from both residues are observed in the DQ-HN(CA) in the absence of HN(CO)(HN(COCA) signals, the identity of 2-13C i−1–13C/15N i pairs is established nevertheless. Because labeled isoleucine is present in one sample only, no distinction between the two 2-13C-labeled residue types in position i − 1 is possible at this stage. A similar situation is met for V20 which is 15N-labeled in sample 3 and preceded by a 2-13C-labeled residue, as evidenced by a cross peak in the CO-filtered HN(CA), but the discrimination between Ala–Val and Tyr–Val pairs remains ambiguous. In contrast, the presence of a HN(COCA) cross peak for V85 unambiguously indicates that this residue is preceded by a leucine. Finally, cross peaks due to L35 are observed in all spectra of sample 3 except for the CO-filtered HN(CA), revealing that it corresponds to the C-terminal leucine residue of a Leu–Leu pair.

Combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling applied to TMD0. Shown in a, b are two different expansions from the full set of 2D 1H–15N correlation spectra recorded on the three samples detailed in Table 1. The indicated sequential assignments were either derived directly from these spectra or required additional information from the 3D HNCA. The cross peak due to V68 in the CO-filtered HN(CA) of sample 3, labeled with a dagger, appears at 13C natural abundance. An empty box in the CO-filtered HN(CA) spectrum of sample 1 indicates the position of I24, which can potentially be observed here, but has insufficient signal-to-noise
The region shown in Fig. 4b comprises signals from a total of 20 residues, eight of which (L11, W51, V93, F120, I125, A129, L132, L136) only appear in HSQC spectra (15N-labeled amino acids) or in HSQC and HNCA spectra (13C/15N-labeled amino acids), yielding the amino acid type but no sequential information. Six residues (V7, F13, V26, F27, A90, A97) give rise to cross peaks in CO-filtered HN(CA) but not in DQ-HN(CA) spectra, reflecting 2-13C i−1-15N i pairs. Of the latter, F13 is observed in the CO-filtered HN(CA) of sample 1 and F27 in those of samples 1 and 2, establishing valine and alanine, respectively, as the preceding residue types. Both combinations are unique in the amino acid sequence of TMD0. For W135 and L137, correlations are obtained in all but CO-filtered HN(CA) spectra, identifying themselves as Trp–Trp and Leu–Leu pairs, respectively, of which the former is unique in the sequence. The two Ile–Tyr pairs in TMD0 are spotted by virtue of cross peaks in the HN(COCA) spectrum of sample 1. Subsequent sequential assignment requires 13Cα chemical shift information taken from the 3D HNCA. Detection of a HN(CO) cross peak for I18 allows for two amino-acid type pairs, Asp–Ile and Pro–Ile, whereas the unique Pro–Arg pair involving R77 is unambiguously identified by the presence and absence of HN(CO) peak for samples 1 and 3, respectively.
With only one exception, all cross peaks expected on the basis of the respective labeling pattern in each of the samples were in fact observed. The full spectra obtained in HSQC and all 2D triple-resonance experiments, including the controls of the CO-filtered HN(CA), are presented in the Supplementary material. The exception is the W115 correlation missing in the control spectrum of the CO-filtered HN(CA) of sample 2. Therefore it cannot be decided with the help of the CO-filtered HN(CA) experiment whether the weak correlation observed in HSQC and HN(CA) spectra corresponds to a 13C/15N-labeled residue or a 15N-labeled one preceded by a 2-13C-labeled amino acid, potentially leading to some confusion. Here, the latter option could be ruled out by the presence of a cross peak in the HN(CO). In cases where a correlation due to a 2-13C i−1–15N i pair is not observed in the CO-filtered HN(CA) for sensitivity reasons, the absence of a cross peak in the control spectrum would prevent it from being misassigned to a 13C/15N-labeled residue. In the current application to TMD0 all such pairs were, however, detected in the CO-filtered HN(CA). Of the eight 2-13C i−1–13C/15N i pairs in TMD0, five (L55, L60, L74, I86, L91) give rise to relatively weak cross peaks in the CO-filtered HN(CA) and strong cross peaks in the control spectrum. Those missing in the CO-filtered HN(CA) (I24, L119, W122) were readily identified by their presence/absence in DQ-HN(CA)/HN(COCA) spectra. It should be noted that edited subspectra containing only signals from 13C/15N-labeled amino acids not preceded by 2-13C-labeled ones can be obtained by subtraction of CO-filtered HN(CA) and the corresponding control spectra (Masterson et al. 2008; Tonelli et al. 2007). However, since this is accompanied by a decrease of signal-to-noise ratios by a factor of 21/2, the procedure is recommended only in cases of abundant sensitivity.
Interplay of combinatorial 15N, 13Cα, 13C′ labeling and 3D HNCA experiments
The information that could be extracted from combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling of TMD0 is summarized in Fig. 5. Residues for which amino-acid type, sequential pairs, and sequence-specific assignments were determined are color-coded in the primary structure of the protein in Fig. 5a. This information is mapped onto the 15N-HSQC spectrum of a uniformly labeled sample of TMD0 in Fig. 5b. The refined protocol provided the amino acid type for 107 of the 138 non-proline residues of TMD0 (77.5 %). Sequential information was obtained for 65 sites (47 %), where 49 pairs (35.5 %) were determined unambiguously and the remaining ones allowed for two different amino acid types at the i − 1 position. The latter ambiguity potentially arises for the three 13C/15N-labeled amino acid types (Ile, Trp, Leu) and the three 15N-labeled amino acid types that are included in only one of the samples (Tyr, Ala, Val), preventing a distinction between the two respective 1-13C- or 2-13C-labeled amino acid types in a combinatorial manner. In the present application this was the case for the combinations Asp/Pro–Ile, Ala/Val–Ile, Pro/Ser–Trp, Tyr/Val–Ala, Asp/Ser–Leu, and Ala/Tyr–Val, (where x/y–z denotes an amino acid z preceded by either x or y), while no ambiguity was encountered for Tyr/Val–Trp, Pro/Ser–Ala, Ala/Tyr–Leu, and Asp/Ser–Val because only one of the two indistinguishable pairs occur in the sequence of TMD0, and none of the tyrosines is preceded by any 1-13C or 2-13C-labeled amino acid type. For two of the 15N-labeled arginine residues (R78 and R109) sequential information was obtained although they are preceded by non-13C-labeled residues and therefore only detected in HSQC spectra. Because the preceding residue types of all other arginines were known from the appearance of cross peaks in the 2D triple-resonance spectra and both R78 and R109 are preceded by the same residue type (i.e. another arginine) the identity of these Arg–Arg pairs was unambiguously established. A total of 19 of the identified sequential pairs (13.8 %) is unique in TMD0, immediately resulting in sequence-specific assignments. This includes one assignment (K2) that was again derived by exclusion, since each of the remaining three lysines is part of a unique dipeptide (vide supra) that could be discerned with the present labeling scheme.

Overview of combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling aided assignment of TMD0. Residues for which the amino acid type could be determined are highlighted in blue in the protein’s amino acid sequence (a). Residues for which additional sequential information became available are marked in green or magenta, where green indicates unambiguous pairs and magenta indicates sequential pairs for which two possibilities exist for the amino acid type in position i − 1. Sequence-specific assignments resulting from identified unique pairs (except for K2, see text) are indicated in red. No information from 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling was available for residues drawn in black. Prolines and the single unassigned residue (D113) are printed in grey. b Assigned [15N, 1H]-BEST-TROSY spectrum (950 MHz) of [U-13C; 15N]-labeled TMD0 using the same color coding as in part a. The expansion showing the most crowded region is plotted at a slightly higher level than the full spectrum. Signals originating from tryptophan and asparagine or glutamine side chains are annotated with Wsc and N/Qsc, respectively. Cross peaks labeled “H” are tentatively assigned to the C-terminal His10 tag
As the next step, these sequential assignments are used as anchor points for analysis of the 3D HNCA spectrum. The longest stretches in the amino acid sequence of TMD0, for which no information about either amino acid types or sequential pairs was available from combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling, comprise only two residues (Fig. 5a). These gaps could be filled in a straightforward manner with the 3D HNCA, finally resulting in nearly complete backbone amide assignment, leaving a single unassigned residue (D113).
It should be recognized that the flow of information between the set of 2D [15N, 1H]-correlation spectra of the selectively labeled samples and the 3D HNCA of the uniformly labeled sample is not unidirectional. Rather, knowledge of 13Cα chemical shifts taken from the latter can be employed to increase the information content of the selective labeling scheme (Shi et al. 2004). An obvious case is glycine, whose amino acid type is almost invariably diagnosed from its highfield 13Cα chemical shift, not only allowing its own identification in [15N, 1H]-correlation maps, but adding information for those sequentially following amides that are detected in the selectively labeled samples. Second, the above mentioned ambiguity concerning the two 1-13C- and 2-13C-labeled residues present in each sample can potentially be resolved, depending on the chosen combination of amino acid types. Among the 1-13C-labeled residue types selected here, serine and proline cannot be reliably distinguished on the basis of their 13Cα chemical shifts, but the 13Cα chemical shift range observed for aspartates in TMD0 does not overlap with those of either serine or proline. As a result, the ambiguous pairs Asp/Pro–Ile and Asp/Ser–Leu are converted into unambiguous sequential pairs, provided that the corresponding interresidual cross peaks are observed in the 3D HNCA. In an analogous manner, ambiguous pairs involving 2-13C-labeled alanine, i.e. Ala/Val–Ile and Ala/Tyr–Val are differentiated based on chemical shifts. A third example involves the amino acid type tyrosine, which is 15N-labeled in one sample only. While the two Ile–Tyr pairs are identified by HN(COCA) correlations, no sequential information is available per se for the remaining three as they are preceded by unlabeled amino acid types in the same sample, i.e. leucine in the case of Y4 and Y56, and threonine in the case of Y124. Because leucines and threonines are unequivocally distinguished by their 13Cα chemical shifts, the combination of 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \)-selective labeling and the 3D HNCA on uniformly labeled protein yields two more sequential pairs (Leu–Tyr) and the sequence-specific assignment of Y124 instead of merely the amino acid type. Ancillary data of the latter type is primarily expected for residue types with a relatively low abundance in a given protein sequence. Taken together, before actually matching intra- and interresidual correlations in the 3D HNCA, classification according to the 13Cα chemical shift range increased the number of known amino acid types, unambiguous sequential pairs, and sequence-specific assignments in TMD0 to 112 (81 %), 67 (48.5 %), and 27 (19.5 %), respectively. It should be noted, however, that in other proteins, that have a larger dispersion of 13Cα chemical shifts, the distinction of certain pairs of residues types may be less straightforward. On the other hand, it is exactly in situations where the HNCA is the only 3D spectrum of reasonable quality that provides sequential connectivities, and where the 13Cα chemical shift bandwidth is narrow, that combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling is most helpful. This essentially suggests α-helical membrane proteins as potential targets for application of the protocol proposed here.
Aiming at the tertiary structure determination of a membrane protein, backbone resonance assignments represent only the first step, which must be followed by side-chain assignments. In cases as the one assumed here, where 3D HNCACB and HN(CA)CO experiments are too insensitive, pulse sequences of the HCC(CO)NH-TOCSY type (Clowes et al. 1993; Grzesiek et al. 1993; Logan et al. 1992; Montelione et al. 1992), which are able to provide side-chain assignments in a convenient manner, will likewise fail. Because the approach presented here yields only 1HN, 15N, and 13Cα chemical shifts, those of the missing backbone nuclei (1Hα and \( ^{13} {\text{C}}^{\prime } \)) must be complemented in the first instance. This can achieved with relative ease using sensitive 3D experiments, i.e. HCACO (Ikura et al. 1990a; Serber et al. 2000) and HNCO (Ikura et al. 1990a; Salzmann et al. 1998). Chemical shift pairs of 1H–13Cα groups can then be used as starting points to correlate side-chain resonances with the backbone via HCCH-COSY (Kay et al. 1990a) or (H)CCH-TOCSY (Bax et al. 1990; Fesik et al. 1990) experiments. Independently, side chain resonances may be assigned with the help of direct NH–CH correlations in 15N and/or 13C edited/separated NOESYs (Ikeya et al. 2011; Lin et al. 2006; Schmidt and Güntert 2013; Xu et al. 2005), where selective labeling is again beneficial to reduce overlap and ambiguities (Hagn and Wagner 2014). As a by-product of combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling, the samples which each contain a single 13C/15N-labeled amino acid type can be employed to obtain simplified HCCH-COSY/TOCSY and NOESY spectra.
Concluding remarks
Contrary to combinatorial labeling approaches that employ an increased number of samples to enhance the information content (Maslennikov et al. 2010; Parker et al. 2004; Staunton et al. 2006; Wu et al. 2006), the previously published triple-selective labeling method (Löhr et al. 2012) relies on spectroscopic means to identify a larger variety of isotopomeric species. In essence, this strategy shifts efforts from the sample preparation to the data acquisition side. Combinatorial 15N, 13Cα, \( ^{13} {\text{C}}^{\prime } \) labeling, described here, goes one step further in the same direction. While the subset of residues for which amino-acid types can be determined remains identical, including 13Cα-labeled amino acids raises the number of residue pairs from 37 to 65 and the number of sequence-specific assignments from 10 to 19 when applied to TMD0, ignoring additional 3D-HNCA based chemical shift information. The price to be paid is the supplemental carbonyl-filtered HN(CA) experiment, required to distinguish \( ^{13} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \) –15N i –\( ^{12} {\text{C}}_{i}^{\alpha } \) from \( ^{12} {\text{C}}_{i - 1}^{\alpha } \)–\( ^{12} {\text{C}}_{i - 1}^{\prime } \)–15N i –\( ^{13} {\text{C}}_{i}^{\alpha } \)-moieties. In the current application this entailed a significant increase of spectrometer time demands from 84 to 165 h for all three samples, which includes the time for recording both filtered and control spectra. In principle, the latter can be omitted provided that all expected peaks are observed in the filtered spectrum, cutting down the additional time by half. This option obviously does not exist when the two spectra are acquired in an interleaved manner. Alternatively, when the control spectrum shows all expected correlations, the regular 2D HN(CA) experiment becomes redundant, allowing for moderate time savings. Nevertheless, the bottleneck of the method remains the CO-filtered HN(CA) experiment whose pulse sequence contains a delay in the order of 10 ms during which transverse relaxation of 13Cα spins takes place. Based on our experience with TMD0, which has a rotational correlation time of 25 ns, we estimate the upper limit of τc to be around 30 ns, above which accumulation times would get impractically long. Applications to larger systems will require the use of deuterated amino acids.
A three-sample combinatorial labeling scheme involves seven 15N-labeled amino acid types, where three are unique to and three are missing in each one of the samples while one is present in all samples. Rules pertaining to the numbers and combinations of the other labeling classes are less restrictive. In the scheme employed here, each sample contained a single 13C/15N-labeled and two 1-13C- and 2-13C-labeled residue types, respectively, the latter being permutations of a total of three 1-13C- and three 2-13C-labeled amino acids, although many other options are conceivable. Also, selecting those amino acid types for 2-13C labeling that are 15N-labeled in the respective third sample (here: Tyr, Ala, Val, see Table 1) is not mandatory. However, it must be mentioned that not all amino acid types are commercially available with the 13C label exclusively in the Cα position, posing some limitation on this choice.
The exact combination of labeled amino acids which maximizes the assignment information for a given protein depends on the individual primary sequence and may be evaluated using software described in the literature (Hefke et al. 2010; Maslennikov et al. 2010). As a general guideline, increasing the number of amino acid types that contain 13C labels yields a higher coverage of residues for which sequential information is obtained, but concomitantly reduces specificity, i.e. the fraction of unambiguous sequential pairs. It should also be noted that supplementing fully 13C/15N labeled amino acids gives rise to additional 1H–15N correlations and therefore provides more residue type information but, on the other hand, gradually diminishes the resolution gain afforded by combinatorial selective labeling methods which is critical to the success of assigning α-helical membrane proteins.
References
Bax A, Ikura M (1991) An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J Biomol NMR 1:99–104
Bax A, Clore GM, Gronenborn AM (1990) 1H–1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J Magn Reson 88:425–431. doi:10.1016/0022-2364(90)90202-K
Bayrhuber M, Riek R (2011) Very simple combination of TROSY, CRINEPT and multiple quantum coherence for signal enhancement in an HN(CO)CA experiment for large proteins. J Magn Reson 209:310–314
Boyd J, Soffe N (1989) Selective excitation by pulse shaping combined with phase modulation. J Magn Reson 85:406–413
Butterwick JA, MacKinnon R (2010) Solution structure and phospholipid interactions of the isolated voltage-sensor domain from KvAP. J Mol Biol 403:591–606
Clowes RT, Boucher W, Hardman CH, Domaille PJ, Laue ED (1993) A 4D HCC(CO)NNH experiment for the correlation of aliphatic side-chain and backbone resonances in 13C/15N labeled proteins. J Biomol NMR 3:349–354. doi:10.1007/Bf00212520
Clubb RT, Thanabal V, Wagner G (1992) A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C′ chemical shifts in 15N–13C-labelled proteins. J Magn Reson 97:213–217
Czisch M, Boelens R (1998) Sensitivity enhancement in the TROSY experiment. J Magn Reson 134:158–160
Demirel O, Bangert I, Tampe R, Abele R (2010) Tuning the cellular trafficking of the lysosomal peptide transporter TAPL by its N-terminal domain. Traffic 11:383–393. doi:10.1111/j.1600-0854.2009.01021.x
Demirel O, Jan I, Wolters D, Blanz J, Saftig P, Tampe R, Abele R (2012) The lysosomal polypeptide transporter TAPL is stabilized by interaction with LAMP-1 and LAMP-2. J Cell Sci 125:4230–4240. doi:10.1242/jcs.087346
Dötsch V, Oswald RE, Wagner G (1996) Amino-acid-type-selective triple-resonance experiments. J Magn Reson 110:107–111
Farjon J, Boisbouvier J, Schanda P, Pardi A, Simorre JP, Brutscher B (2009) Longitudinal-relaxation-enhanced NMR experiments for the study of nucleic acids in solution. J Am Chem Soc 131:8571–8577. doi:10.1021/ja901633y
Favier A, Brutscher B (2011) Recovering lost magnetization: polarization enhancement in biomolecular NMR. J Biomol NMR 49:9–15
Fesik SW, Eaton HL, Olejniczak ET, Zuiderweg ERP, Mcintosh LP, Dahlquist FW (1990) 2D and 3D NMR-spectroscopy employing 13C–13C magnetization transfer by isotropic mixing—Spin system-identification in large proteins. J Am Chem Soc 112:886–888. doi:10.1021/Ja00158a069
Feuerstein S, Plevin MJ, Willbold D, Brutscher B (2012) iHADAMAC: a complementary tool for sequential resonance assignment of globular and highly disordered proteins. J Magn Reson 214:329–334. doi:10.1016/j.jmr.2011.10.019
Geen H, Freeman R (1991) Band-selective radiofrequency pulses. J Magn Reson 93:93–141
Gossert AD, Hinniger A, Gutmann S, Jahnke W, Strauss A, Fernández C (2011) A simple protocol for amino acid type selective isotope labeling in insect cells with improved yields and high reproducibility. J Biomol NMR 51:449–456. doi:10.1007/s10858-011-9570-9
Griffey RH, Redfield AG, Loomis RE, Dahlquist FW (1985) Nuclear magnetic resonance observation and dynamics of specific amide protons in T4 lysozyme. Biochemistry 24:817–822
Griffey RH, Redfield AG, Mcintosh LP, Oas TG, Dahlquist FW (1986) Assignment of proton amide resonances of T4 lysozyme by 13C and 15N multiple isotopic labeling. J Am Chem Soc 108:6816–6817. doi:10.1021/Ja00281a066
Grzesiek S, Bax A (1993) Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J Biomol NMR 3:185–204
Grzesiek S, Anglister J, Bax A (1993) Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Magn Reson, Ser B 101:114–119. doi:10.1006/jmrb.1993.1019
Hagn F, Wagner G (2014) Structure refinement and membrane positioning of selectively labeled OmpX in phospholipid nanodiscs. J Biomol NMR. doi:10.1007/s10858-014-9883-6
Hefke F, Bagaria A, Reckel S, Ullrich SJ, Dötsch V, Glaubitz C, Güntert P (2010) Optimization of amino acid type-specific 13C and 15N labeling for the backbone assignment of membrane proteins by solution- and solid-state NMR with the UPLABEL algorithm. J Biomol NMR 49:75–84
Ikeya T et al (2011) Exclusively NOESY-based automated NMR assignment and structure determination of proteins. J Biomol NMR 50:137–146. doi:10.1007/s10858-011-9502-8
Ikura M, Kay LE, Bax A (1990a) A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 29:4659–4667
Ikura M, Krinks M, Torchia DA, Bax A (1990b) An efficient NMR approach for obtaining sequence-specific resonance assignments of larger proteins based on multiple isotopic labeling. FEBS Lett 266:155–158
Jaipuria G, Krishnarjuna B, Mondal S, Dubey A, Atreya HS (2012) Amino acid selective labeling and unlabeling for protein resonance assignments. Adv Exp Med Biol 992:95–118. doi:10.1007/978-94-007-4954-2_6
Kainosho M, Tsuji T (1982) Assignment of the three methionyl carbonyl carbon resonances in Streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution. Biochemistry 21:6273–6279
Kay LE, Ikura M, Bax A (1990a) Proton–proton correlation via carbon–carbon couplings: a three-dimensional NMR approach for the assignment of aliphatic resonances in proteins labeled with carbon-13. J Am Chem Soc 112:888–889. doi:10.1021/Ja00158a070
Kay LE, Ikura M, Tschudin R, Bax A (1990b) Three-dimensional triple-resonance NMR-spectroscopy of isotopically enriched proteins. J Magn Reson 89:496–514
Kay L, Keifer P, Saarinen T (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114:10663–10665
Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR (2009) Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Magn Reson Spectrosc 55:335–360
Klammt C et al (2004) High level cell-free expression and specific labeling of integral membrane proteins. Eur J Biochem 271:568–580
Kupče E, Freeman R (1994) Wide-band excitation with polychromatic pulses. J Magn Reson A 108:268–273
Kupče E, Boyd J, Campbell ID (1995) Short selective pulses for biochemical applications. J Magn Reson, Ser B 106:300–303. doi:10.1006/jmrb.1995.1049
Lee D, Hilty C, Wider G, Wüthrich K (2006) Effective rotational correlation times of proteins from NMR relaxation interference. J Magn Reson 178:72–76
LeMaster DM, Richards FM (1985) 1H–15N heteronuclear NMR studies of Escherichia coli thioredoxin in samples isotopically labeled by residue type. Biochemistry 24:7263–7268
Lescop E, Rasia R, Brutscher B (2008) Hadamard amino-acid-type edited NMR experiment for fast protein resonance assignment. J Am Chem Soc 130:5014–5015
Lescop E, Kern T, Brutscher B (2010) Guidelines for the use of band-selective radiofrequency pulses in hetero-nuclear NMR: example of longitudinal-relaxation-enhanced BEST-type 1H–15N correlation experiments. J Magn Reson 203:190–198. doi:10.1016/j.jmr.2009.12.001
Lin Z, Xu Y, Yang S, Yang D (2006) Sequence-specific assignment of aromatic resonances of uniformly 13C,15N-labeled proteins by using 13C- and 15N-edited NOESY spectra Angewandte Chemie (International ed in English) 45:1960–1963
Logan TM, Olejniczak ET, Xu RX, Fesik SW (1992) Side chain and backbone assignments in isotopically labeled proteins from two heteronuclear triple resonance experiments. FEBS Lett 314:413–418
Löhr F et al (2012) Combinatorial triple-selective labeling as a tool to assist membrane protein backbone resonance assignment. J Biomol NMR 52:197–210. doi:10.1007/s10858-012-9601-1
Maslennikov I, Choe S (2013) Advances in NMR structures of integral membrane proteins. Curr Opin Struct Biol 23:555–562
Maslennikov I et al (2010) Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc Natl Acad Sci USA 107:10902–10907
Masterson LR, Tonelli M, Markley JL, Veglia G (2008) Simultaneous detection and deconvolution of congested NMR spectra containing three isotopically labeled species. J Am Chem Soc 130:7818–7819
McIntosh LP, Dahlquist FW (1990) Biosynthetic incorporation of 15N and 13C for assignment and interpretation of nuclear magnetic resonance spectra of proteins. Q Rev Biophys 23:1–38
Michel E, Skrisovska L, Wüthrich K, Allain FH (2013) Amino acid-selective segmental isotope labeling of multidomain proteins for structural biology. ChemBioChem 14:457–466. doi:10.1002/cbic.201200732
Montelione GT, Wagner G (1990) Conformation-independent sequential NMR connections in isotope-enriched polypeptides by 1H–13C–15N triple-resonance experiments. J Magn Reson 87:183–188. doi:10.1016/0022-2364(90)90098-T
Montelione GT, Lyons BA, Emerson SD, Tashiro M (1992) An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J Am Chem Soc 114:10974–10975. doi:10.1021/Ja00053a051
Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW (1989) Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol 177:44–73
Nietlispach D, Gautier A (2011) Solution NMR studies of polytopic α-helical membrane proteins. Curr Opin Struct Biol 21:497–508
Nietlispach D, Ito Y, Laue ED (2002) A novel approach for the sequential backbone assignment of larger proteins: selective intra-HNCA and DQ-HNCA. J Am Chem Soc 124:11199–11207
Ozawa K, Headlam MJ, Schaeffer PM, Henderson BR, Dixon NE, Otting G (2004) Optimization of an Escherichia coli system for cell-free synthesis of selectively 15N-labelled proteins for rapid analysis by NMR spectroscopy. Eur J Biochem 271:4084–4093. doi:10.1111/j.1432-1033.2004.04346.x
Ozawa K, Wu PS, Dixon NE, Otting G (2006) 15N-Labelled proteins by cell-free protein synthesis. Strategies for high-throughput NMR studies of proteins and protein-ligand complexes. FEBS J 273:4154–4159
Pantoja-Uceda D, Santoro J (2008) Amino acid type identification in NMR spectra of proteins via β- and γ-carbon edited experiments. J Magn Reson 195:187–195. doi:10.1016/j.jmr.2008.09.010
Parker MJ, Aulton-Jones M, Hounslow AM, Craven CJ (2004) A combinatorial selective labeling method for the assignment of backbone amide NMR resonances. J Am Chem Soc 126:5020–5021
Patt SL (1992) Single-frequency-shifted and multiple-frequency-shifted laminar pulses. J Magn Reson 96:94–102
Pervushin K, Riek R, Wider G, Wüthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371
Pervushin KV, Wider G, Wüthrich K (1998) Single transition-to-single transition polarization transfer (ST2-PT) in [15N,1H]-TROSY. J Biomol NMR 12:345–348
Rasia RM, Brutscher B, Plevin MJ (2012) Selective isotopic unlabeling of proteins using metabolic precursors: application to NMR assignment of intrinsically disordered proteins. ChemBioChem 13:732–739. doi:10.1002/cbic.201100678
Reckel S et al (2008) Transmembrane segment enhanced labeling as a tool for the backbone assignment of alpha-helical membrane proteins. Proc Natl Acad Sci USA 105:8262–8267. doi:10.1073/pnas.0710843105
Rios CB, Feng W, Tashiro M, Shang Z, Montelione GT (1996) Phase labeling of C–H and C–C spin-system topologies: application in constant-time PFG-CBCA(CO)NH experiments for discriminating amino acid spin-system types. J Biomol NMR 8:345–350
Rule GS, Tjandra N, Simplaceanu V, Ho C (1993) Assignment strategies for 15N–1H correlated spectra of large proteins in solution. J Magn Reson 102:126–128
Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K (1998) TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA 95:13585–13590
Sanders CR, Sönnichsen F (2006) Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem 44:S24–S40. doi:10.1002/mrc.1816
Schanda P, Van Melckebeke H, Brutscher B (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J Am Chem Soc 128:9042–9043
Schmidt E, Güntert P (2013) Reliability of exclusively NOESY-based automated resonance assignment and structure determination of proteins. J Biomol NMR 57:193–204. doi:10.1007/s10858-013-9779-x
Schubert M, Smalla M, Schmieder P, Oschkinat H (1999) MUSIC in triple-resonance experiments: amino acid type-selective 1H–15N correlations. J Magn Reson 141:34–43
Schwarz D et al (2007) Preparative scale expression of membrane proteins in Escherichia coli-based continuous exchange cell-free systems. Nat Protoc 2:2945–2957
Serber Z et al (2000) New carbon-detected protein NMR experiments using CryoProbes. J Am Chem Soc 122:3554–3555. doi:10.1021/Ja991371m
Shenkarev ZO et al (2010) NMR structural and dynamical investigation of the isolated voltage-sensing domain of the potassium channel KvAP: implications for voltage gating. J Am Chem Soc 132:5630–5637
Shi J, Pelton JG, Cho HS, Wemmer DE (2004) Protein signal assignments using specific labeling and cell-free synthesis. J Biomol NMR 28:235–247
Shortle D (1994) Assignment of amino acid type in 1H–15N correlation spectra by labeling with 14N-amino acids. J Magn Reson 105:88–90
Smith MA, Hu H, Shaka AJ (2001) Improved broadband inversion performance for NMR in liquids. J Magn Reson 151:269–283. doi:10.1006/jmre.2001.2364
Sobhanifar S et al (2010) Cell-free expression and stable isotope labelling strategies for membrane proteins. J Biomol NMR 46:33–43
Staunton D, Schlinkert R, Zanetti G, Colebrook SA, Campbell ID (2006) Cell-free expression and selective isotope labelling in protein NMR. Magn Reson Chem 44:S2–S9. doi:10.1002/mrc.1835
Takeuchi K, Ng E, Malia TJ, Wagner G (2007) 1-13C amino acid selective labeling in a 2H15N background for NMR studies of large proteins. J Biomol NMR 38:89–98. doi:10.1007/s10858-007-9152-z
Tamm LK, Liang B (2006) NMR of membrane proteins in solution. Prog Nucl Magn Reson Spectrosc 48:201–210
Tate S et al (1992) Stable isotope aided nuclear magnetic resonance study to investigate the receptor-binding site of human interleukin 1β. Biochemistry 31:2435–2442
Tonelli M, Masterson LR, Hallenga K, Veglia G, Markley JL (2007) Carbonyl carbon label selective (CCLS) 1H–15N HSQC experiment for improved detection of backbone 13C–15N cross peaks in larger proteins. J Biomol NMR 39:177–185. doi:10.1007/s10858-007-9185-3
Trbovic N, Klammt C, Koglin A, Löhr F, Bernhard F, Dötsch V (2005) Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc 127:13504–13505
Tumulka F, Roos C, Löhr F, Bock C, Bernhard F, Dötsch V, Abele R (2013) Conformational stabilization of the membrane embedded targeting domain of the lysosomal peptide transporter TAPL for solution NMR. J Biomol NMR 57:141–154. doi:10.1007/s10858-013-9774-2
Vajpai N, Strauss A, Fendrich G, Cowan-Jacob SW, Manley PW, Grzesiek S, Jahnke W (2008a) Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J Biol Chem 283:18292–18302. doi:10.1074/jbc.M801337200
Vajpai N, Strauss A, Fendrich G, Cowan-Jacob SW, Manley PW, Jahnke W, Grzesiek S (2008b) Backbone NMR resonance assignment of the Abelson kinase domain in complex with imatinib. Biomol NMR Assign 2:41–42. doi:10.1007/s12104-008-9079-7
Weigelt J (1998) Single scan, sensitivity- and gradient-enhanced TROSY for multidimensional NMR Experiments. J Am Chem Soc 120:10778–10779
Weigelt J, van Dongen M, Uppenberg J, Schultz J, Wikström M (2002) Site-selective screening by NMR spectroscopy with labeled amino acid pairs. J Am Chem Soc 124:2446–2447
Wishart DS et al (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140
Wittekind M, Mueller L (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson 101:201–205
Wu PS, Ozawa K, Jergic S, Su XC, Dixon NE, Otting G (2006) Amino-acid type identification in 15N-HSQC spectra by combinatorial selective 15N-labelling. J Biomol NMR 34:13–21
Xu Y, Lin Z, Ho C, Yang D (2005) A general strategy for the assignment of aliphatic side-chain resonances of uniformly 13C,15N-labeled large proteins. J Am Chem Soc 127:11920–11921
Yabuki T et al (1998) Dual amino acid-selective and site-directed stable-isotope labeling of the human c-Ha-Ras protein by cell-free synthesis. J Biomol NMR 11:295–306
Acknowledgments
This work was supported by the state of Hesse (Center for Biomolecular Magnetic Resonance), the German Research Foundation (SFB 807), NIH (U54GM087519) and the Cluster of Excellence Frankfurt (Macromolecular Complexes).
Conflict of interest
The authors declare that they have no conflict of interest. This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Löhr, F., Tumulka, F., Bock, C. et al. An extended combinatorial 15N, 13Cα, and \( ^{13} {\text{C}}^{\prime } \) labeling approach to protein backbone resonance assignment. J Biomol NMR 62, 263–279 (2015). https://doi.org/10.1007/s10858-015-9941-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-015-9941-8