
Abstract
We present a computational method for finding optimal labeling patterns for the backbone assignment of membrane proteins and other large proteins that cannot be assigned by conventional strategies. Following the approach of Kainosho and Tsuji (Biochemistry 21:6273–6279 (1982)), types of amino acids are labeled with 13C or/and 15N such that cross peaks between 13CO(i – 1) and 15NH(i) result only for pairs of sequentially adjacent amino acids of which the first is labeled with 13C and the second with 15N. In this way, unambiguous sequence-specific assignments can be obtained for unique pairs of amino acids that occur exactly once in the sequence of the protein. To be practical, it is crucial to limit the number of differently labeled protein samples that have to be prepared while obtaining an optimal extent of labeled unique amino acid pairs. Our computer algorithm UPLABEL for optimal unique pair labeling, implemented in the program CYANA and in a standalone program, and also available through a web portal, uses combinatorial optimization to find for a given amino acid sequence labeling patterns that maximize the number of unique pair assignments with a minimal number of differently labeled protein samples. Various auxiliary conditions, including labeled amino acid availability and price, previously known partial assignments, and sequence regions of particular interest can be taken into account when determining optimal amino acid type-specific labeling patterns. The method is illustrated for the assignment of the human G-protein coupled receptor bradykinin B2 (B2R) and applied as a starting point for the backbone assignment of the membrane protein proteorhodopsin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The structure determination of membrane proteins, amyloid fibrils, and large protein complexes by either solution or solid-state NMR is a challenging task. While structure determination by solid-state NMR has been successful for small model proteins, i.e. the α-spectrin SH3 domain (Castellani et al. 2002), ubiquitin (Zech et al. 2005), and the immunoglobulin-binding B1 domain of the streptococcal protein G (GB1) (Franks et al. 2008; Zhou et al. 2007), further methodological advances are required to extend the applicability of this technique to larger systems of biological and pharmacological interest, e.g. G protein-coupled receptors or ribosomal complexes. Recent developments in labeling strategies, NMR hardware, pulse sequences and structure determination protocols suggest that the study of such large systems is feasible. With regard to the structural analysis of helical membrane proteins by NMR spectroscopy, the difficulty of obtaining sufficient amounts of purified protein, homogenously reconstituted in an appropriate hydrophobic environment is a first limitation to overcome. Furthermore, the analysis of NMR spectra of membrane proteins is hampered by the low chemical shift dispersion of the helical secondary structure, resulting in an overall low sensitivity and a high degree of signal overlap. In addition, in solution NMR the size of the proteo-micelle leads to broad lines due to slow tumbling and fast relaxation which further aggravates signal overlap and low sensitivity. Thus, standard labeling techniques and experiments designed for the backbone assignment in solution as well as in the solid state are only partially successful and selective labeling becomes an indispensable tool. In this context, a suite of labeling and assignment strategies can help to obtain sequence-specific resonance assignments for proteins with poor resolution.
A widely-used labeling scheme for the sequential assignment and the measurement of long-range distance restraints by solid-state NMR for use in structure calculations is based on the usage of [1,3-13C]- and [2-13C]-glycerol (Higman et al. 2009). Other labeling schemes used to obtain resonance assignments in the solid state include the use of [1,4-13C], [2,3-13C], and [1,2,3,4-13C] succinic acid based samples (van Gammeren et al. 2004) and reverse labeling in which key hydrophobic residues remain unlabeled (Etzkorn et al. 2007; Schneider et al. 2008). Other labeling schemes, such as fractional [U-13C]-glucose labeling (Schubert et al. 2006) or labeling with [1-13C]-glucose (Hong 1999) or [2-13C]-glucose (Lundström et al. 2007) provide alternative labeling approaches that may be well suited depending on protein secondary structure and amino acid type composition.
In solution NMR, segmental labeling has been employed to incorporate stable isotopes into one or several sequence regions of a protein (Busche et al. 2009; Skrisovska and Allain 2008; Yamazaki et al. 1998; Züger and Iwai 2005). Transmembrane segment enhanced labeling (Reckel et al. 2008) is a tool for the backbone assignment of α-helical membrane proteins that relies on the fact that approximately 60% of the amino acids in transmembrane regions consist of only six different amino acid types, Ala, Gly, Ile, Leu, Phe, and Val. By 15N/13C-double-labeling of these amino acid types, sequential connectivities can be obtained for stretches composed exclusively of these six amino acid types as they occur predominantly in the transmembrane segments. Stereo-array isotope labeling (SAIL) (Kainosho and Güntert 2009; Kainosho et al. 2006), which applies a complete stereospecific and regiospecific labeling pattern that is optimal with regard to the quality and information content of the resulting NMR spectra, is a further approach to reduce relaxation and overlap, in particular for the side-chain atoms.
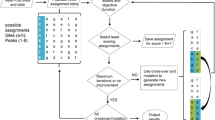
However, with increasing line-widths and complexity of the spectra most of these labeling and assignment strategies become unfeasible. In order to address this problem, we present here a computer algorithm that enables the optimal use of an assignment method based upon amino acids labeled selectively with 13C or/and 15N (Kainosho and Tsuji 1982). This method can unambiguously assign the polypeptide backbone of unique pairs of adjacent amino acids in the protein sequence. Unique pairs are distinguished from each other by a combination of samples with different amino acid type-specific labeling patterns as has been described previously (Maslennikov et al. 2010; Parker et al. 2004; Shi et al. 2004; Trbovic et al. 2005). The low density of NMR-active nuclei in such samples gives rise to higher quality spectra than with uniformly 13C/15N-labeled protein samples. A given amino acid pair at the sequence positions i−1 and i gives rise to a cross peak between 13CO(i – 1) and 15NH(i) in a HNCO, HN(CO), or NCO spectrum if and only if the first residue is labeled with 13C and the second with 15N. In principle, all unique pairs of amino acids in a protein can be assigned with this approach by preparing for each unique pair a sample in which the first residue type is labeled with 13C and the second with 15N. However, the high number of samples that would have to be prepared and measured by NMR makes this brute force approach unpractical. Instead, the same result can be achieved with a far smaller number of differently labeled protein samples by exploiting the peak presence and absence patterns of a given amino acid pair in samples with multiple 13C or/and 15N labeled amino acid types (Maslennikov et al. 2010; Parker et al. 2004; Trbovic et al. 2005). The purpose of our new algorithm is to determine optimal labeling patterns by combinatorial optimization under a variety of auxiliary conditions that are important for practical applications (Fig. 1).

Input and output data of the UPLABEL algorithm
Materials and methods
Definitions
Two amino acids residues form an (ordered) pair if they are next to each other in the protein sequence. A pair is a unique pair if no other pair in the protein sequence is composed of the same amino acid types. We denote the set of all pairs as AA, and the subset of all unique pairs of interest as UP. A labeling scheme with n samples consists of n subsets U 1,…, U n ⊆ AA of labeled pairs in which the first amino acid is labeled with 13C and the second with 15N. A labeled pair gives rise to a peak in a HNCO, HN(CO), or NCO spectrum. Two labeled pairs can lead to the co-occurrence of further labeled pairs. For instance, in the sequence Gly-Thr-Ala-Thr-Ala-Gly the labeling of the pairs Gly-Thr and Ala-Gly requires 13C,15N-Gly, 13C-Ala, and 15N-Thr, which results in the co-labeling of the pair Ala-Thr. The peak occurrence pattern of a pair is the set of spectra that contain a peak for the given pair. A peak occurrence pattern is uniquely encoded into its pattern number, given by the integer number in which the j-th bit is set if and only if the pair is labeled in the sample j = 1, …, n. An unambiguous sequence-specific resonance assignment of a unique pair can be obtained if its pattern number is unique, i.e., if no other pair gives rise to the same peak pattern. The maximal pattern number, and thus the maximal number of pairs that can be assigned by a labeling scheme, is 2n−1. However, co-labeling between pairs can limit the number of amino acids that can be added to a sample, because the peak occurrence pattern needs to be unique for an unambiguous assignment. A labeling scheme is optimal if it yields, under given auxiliary conditions, the maximal number of (or, according to a scoring function, the most valuable) unambiguous assignments using the smallest number of samples.
Graph representation of the problem
The problem to find an optimal combinatorial labeling scheme belongs to the class of NP-hard problems which are difficult to solve because the computation time to find an exact solution increases exponentially with the problem size. To show this, the problem is transformed into a graph problem. Unique pairs are represented by nodes x ∈ UP, and an edge exists between two nodes x 1 and x 2 if their labeling leads to the co-labeling of a third pair x 3 ∈ AA. A special case of the problem is that the labeling scheme can only consist of unique pairs. This can be achieved by modifying the graph so that an edge exists only when two pairs co-label a non-unique pair. This subproblem corresponds to the NP-complete set packing problem (Ausiello et al. 1980) in that every sample corresponds to a set packing. Hence, the general problem must also belong to the class of NP-complete problems.
The UPLABEL algorithm
Our UPLABEL algorithm for finding (approximately) optimal labeling schemes is implemented in the CYANA program package (Güntert 1997), in a freely available standalone program, and by a web portal at http://www.bpc.uni-frankfurt.de/guentert/wiki/index.php/UPLABEL. It employs a randomized greedy algorithm to generate labeling schemes that can be scored afterwards. The general strategy is to transform a sequence of unique amino acid pairs x 1,…, x N ∈ UP into a labeling scheme that carries as much information as possible, as expressed by the score (see below). This preliminary solution is then improved by permuting the order of the input pairs x 1,…, x N and repeating the scoring until convergence is reached or a user-defined time limit is exceeded. As in a dynamic algorithm, subsolutions are stored and used to speed up the computation. The algorithm uses several iterations with randomized input to reduce the risk of encountering a bad solution.
First all N unique pairs x 1,…, x N ∈ UP are assembled from the amino acid sequence of the protein and a matrix is set up that stores the information whether the labeling of two pairs x 1 ∈ AA and x 2 ∈ AA leads to the co-labeling of a third pair x 3 ∈ AA. The unique pairs are then added to the labeling scheme in the order described by x 1,…, x N . The pair is assigned greedily to the pattern number that maximizes the scoring function for that pair, while preserving the uniqueness of all pattern numbers of unique pairs and user defined properties. Whenever a pair cannot be added to the labeling scheme with the current number of samples, a new sample is added. Following this iterative procedure the ordered set of unique pairs x 1,…, x N is mapped to pattern numbers in the range 1, …, 2n−1, which establishes a labeling scheme with n samples. Repeating the procedure with different initial permutations of the unique pairs leads to a sampling of different labeling schemes. The ten distinct labeling schemes with the best score are saved.
Finding a favorable sampling scheme iteratively allows the algorithm to discard partial labeling schemes that have no chance to be optimal. Because the addition of a new sample doubles the number of available pattern numbers, the searching time for a pattern number also doubles with every extra sample. Cutting off at a certain pattern number thus saves computing time, excluding unique pairs that cannot be assigned using the number of samples.
Saving partial solutions is another way to reduce the computation time. Partial labeling schemes consisting of i < N unique pairs are stored in a tree. The path from the root of the tree to the leaves is formed by the permutation of unique pairs. Each node stores the partial labeling scheme that consists of the pairs x 1,…, x i . Another child node is then appended, extending the labeling scheme with a new pair x i+1. This allows re-using part of an already completed labeling scheme as the starting point for the addition of new pairs. A new labeling scheme is computed with the next permutation. This technique is similar to using a dynamic programming algorithm, but allows for more flexibility in the order the permutations are tested.
Since the number of possible permutations is prohibitively large for a larger number of unique pairs, an alternative strategy was also implemented. This second algorithm first designs an approximate labeling scheme that does not necessarily preserve the uniqueness of pattern numbers and then locally optimizes it by removing duplicate occurrence patterns. If possible, the removed pairs are again added to the labeling scheme with a different pattern number. Since the number of amino acids pairs affected by removing a unique pair from a sample decreases with a growing number of samples the effectiveness of this approach rises with a growing number of samples. This second algorithm works especially well if the number of samples is greater than the dyadic logarithm of the number of unique pairs because most pairs can then be integrated into the labeling scheme, while the first algorithm is better at deciding which pairs to exclude. Therefore, in an UPLABEL calculation the second algorithm is used to create a labeling scheme that serves as a lower bound for the first algorithm so that convergence is reached sooner.
The scoring function
The scoring function measures desirable properties of a (partial or complete) labeling scheme. The UPLABEL algorithm uses the scoring function to implement restraints which are used to select optimal labeling schemes among all labeling schemes that are compatible with the constraint of uniqueness of the peak occurrence patterns.
In the present implementation of the UPLABEL algorithm, the scoring function F is the weighted sum of four terms that capture different aspects of a labeling scheme, Λ,
The individual terms in the scoring function are normalized to values between 0 and 1 by scaling with the values for the theoretical best and worst labeling schemes. In addition, every term carries a user-defined weighting factor through which the labeling scheme can be tuned by the user.
The first term of the score is inversely proportional to the number of samples used, n, and normalized by log2 N, the minimal number of samples theoretically needed if there were no pairs that co-label other pairs:
The second term takes into account the “price” of labeled amino acids. Given prices P(a,γ) for the standard amino acid types a = 1, …, 20 labeled with isotope(s) γ = 13C, 15N, or 13C/15N, the contribution to the score is given by
S denotes the number of samples, and δ(a,s,γ) equals 1 if the amino acid type a is labeled in sample s = 1, …, S with the isotope(s) γ, and 0 otherwise. The total cost of the labeling scheme is normalized by the maximal total cost that would be incurred by using in all samples the most expensive labeling for each amino acid.
The third term measures the fraction of all available pattern numbers for a given number n of samples that are used for labeling M pairs:
Because of co-labeling or incomplete labeling of the unique pairs, the number of used pattern numbers, M, may be larger or smaller than the number of unique pairs of interest, N.
The fourth term is used to weigh regions of interest with a user-defined interest factor I(l) for all l = 1, …, L amino acid residues of the protein.
The term μ(l,s) equals 1 if the residue l is part of a labeled pair in the sample s, and 0 otherwise.
Auxiliary conditions
The basic optimization problem of finding a labeling scheme that yields unambiguous assignments for as many unique pairs as possible using as few samples as possible needs in practice often be modified because additional conditions have to be met by the labeling scheme. The UPLABEL was thus designed to be flexible with regard to imposing various auxiliary conditions on labeling schemes. If 13C, 15N, and 13C/15N labeling is available only for a subset of the standard amino acids, the labeling scheme can be restricted to include only the available labeled amino acids. A limit on the maximal number of expected peaks in the spectra can be imposed by the user to avoid excessive overlap. To concentrate the assignment effort on given regions of the protein sequence, the set UP of unique pairs can be redefined freely by the user to comprise only a subset of all unique pairs, and/or to include also additional, non-unique pairs. This can be useful if parts of the sequence have already been assigned and the combinatorial labeling serves to fill missing gaps in the assignment. Alternatively, the main biological interest may lie in a certain region of the protein such as the active site, the loop regions, or the transmembrane regions of a membrane protein, while assignments of other parts of the protein are not required. In the first case it is permissible to include also non-unique pairs if all but one occurrences of a pair are already assigned. In the second case labeling schemes will be determined that allow predominantly the assignment of unique pairs in the region of interest. In certain cases it may also be acceptable to get assignments with a user-defined maximal degree of ambiguity because other knowledge may still enable a meaningful interpretation of the results. For instance, if the structure of the protein is known, it can be used to resolve ambiguities.
Amino acid type-specific (un)labeled NMR samples can also be produced at low cost starting from uniformly labeled 13C and 15N sources by specifically adding an excess of unlabeled amino acids. The amino acid residues of the resulting protein will be either doubly 13C/15N-labeled or unlabeled. The UPLABEL algorithm can also be applied in this situation to evaluate whether employing an un-labeling strategy would be feasible. This technique is very similar to the labeling approach proposed by (Parker et al. 2004) that also uses only doubly labeled amino acids. In practice, it can be advantageous to decrease the labeling cost by using a combination of samples with specific un-labeling and normal partial amino acid type-specific labeling.
If several related sequences are to be assigned by combinatorial labeling, the number of labeled amino acid types required can be minimized by keeping the labeling of the common unique pairs of both sequences the same. The remaining unique pairs can then be re-added to get two labeling schemes using largely the same labeled amino acid types.
Editing of labeling schemes
Once good labeling schemes have been found by the UPLABEL algorithm, the user may consider additional criteria that are not incorporated into the scoring function. The UPLABEL algorithm allows the import, manual editing, and optimization of a labeling scheme, which can then be rescored against the built-in scoring function. Samples that can be extended by a certain pair without causing assignment ambiguities are reported by the program. The user can remove pairs or add new pairs, including non-unique pairs, with the algorithm exhaustively trying all permutations of those pairs using an optimization function to remove redundancies and to lower the cost of labeling. The program can provide statistical information on the expected peaks per sample, the average number of expected peaks, and the number of peaks for which a unique assignment is possible. Since the UPLABEL algorithm is integrated into CYANA, general CYANA functions can be used to analyze expected properties of a sample, for example, the expected extent of peak overlap (Schmucki 2008; Schmucki et al. 2009).
Input and output
The input data comprise necessary and optional parameters (Fig. 1). In its basic form, the UPLABEL algorithm requires as input only the amino acid sequence of the protein. However, to find optimal labeling schemes in various commonly occurring situations, the user may provide additional information. Amino acids that are subject to scrambling can be excluded from the labeling scheme. The search for optimal labeling schemes can be further improved by putting emphasis on certain amino acid types or certain regions of the sequence to take into account the availability of amino acid types and/or already gathered information, such as the amino acid type specific “prices” of Eq. (3) that allow the exclusive or preferential use of certain labeled amino acid types (Maslennikov et al. 2010), and specifying the importance of assigning certain sequence positions through Eq. (5). Optionally, also duplicate patterns or amino acid pairs that occur multiply in the sequence can be included in labeling schemes, e.g. if they may be assigned by other, complementary methods. The “marker” parameter can be used to add an extra dimension to the labeling scheme, if a peak cannot be distinguished by the labeling scheme alone, but there are experiments available to distinguish it by other means. A limit on the maximal number of peaks per sample can be set by the user to reduce possible spectral overlap, if necessary.
To facilitate examining the results of the computation, the (typically ten) best-scoring labeling schemes can be visualized in different ways. The “samples presentation” lists the samples with their proposed composition of 15N, 13C, and 13C,15N-labeled amino acids. This presentation is useful for the preparation of the samples and for comparing labeling schemes. The “sequence presentation” shows the pairs together with their respective labeling patterns along the sequence. This view is used to get an overview of the pairs that can be assigned using a labeling scheme, and their respective peak patterns. Examples are shown in the Results section.
Results and discussion
Our goal was to create a flexible software package that is capable of finding labeling schemes for the backbone assignment of membrane proteins and other proteins that are difficult to assign by traditional methods. Here we show results obtained by the UPLABEL algorithm. Special care was taken to incorporate as far as possible common restrictions and auxiliary conditions when designing labeling schemes and to make the output easily understandable.
Expected number of unique pairs
The probability of a pair to be unique in a protein sequence composed of L amino acid residues can be estimated as (1−p 2)L−2, assuming that all 20 amino acid types occur independently and with equal probability, p = 1/20, at each sequence position (Reckel et al. 2008). One would thus expect 78% unique pairs in a protein of 100 residues, and 47% unique pairs in a protein of 300 residues. Taking into account that the amino acid types occur in proteins with different probabilities p 1, …, p 20 (McCaldon and Argos 1988), the probability of a pair to be unique decreases to \( \sum\nolimits_{i,j = 1}^{20} {p_{i} p_{j} (1 - p_{i} p_{j} )^{L - 2} } \), or 72% and 40% for proteins of 100 and 300 residues, respectively. Even though the distribution of amino acid residue types in membrane proteins is less uniform than in general proteins, this indicates that a significant number of assignments can be achieved by this method. For instance, the human G-protein coupled receptor bradykinin B2 (B2R) with 391 residues contains 115 (29%) unique pairs, excluding proline-containing pairs.
The labeling of multiple unique pairs in a sample can lead to the undesired co-labeling of further pairs. Labeling m unique pairs A 1 B 1, …, A m B m by incorporating 13C into the amino acid types A 1, …, A m and 15N into the amino acid types B 1, …, B m leads to the labeling of all pairs {A i ,B j }, i, j = 1,…, m, that occur in the protein sequence. The probability that only the m intended unique pairs A 1 B 1, …, A m B m (but no others) are labeled is approximately (1−p 2)m(m−2) in the case of uniform amino acid type occurrence, i.e. 82% for m = 10 or 41% for m = 20 unique pairs.
Control application for a sequence with known optimal solution
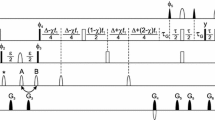
To verify the correct functioning of the UPLABEL algorithm in a case for which an optimal solution is known we applied it to a 16 amino acid residue fragment from the bradykinin receptor B2R (Fig. 2). This sequence comprises 15 pairs, all of which are unique. This is the theoretically maximal number of unique pairs that can be assigned with 4 samples (15 = 24−1). Co-labeling does not impose further restrictions for this sequence. Indeed, the UPLABEL algorithm yielded a labeling scheme that allows the unambiguous assignment of all 15 unique pairs (Fig. 2).

Unique pair labeling of the fragment comprising 170–185 of the human G-protein coupled receptor bradykinin B2 (B2R). The labeling patterns of the pairs in four samples are shown below the sequence, indicating labeled pairs with an ‘X’ between the two amino acids that form a pair. The two rows above the sequence show, respectively, the number of occurrences of amino acid pairs (always ‘1’ since all pairs are unique), and the unique pairs that can be assigned unambiguously (indicated by ‘X’) by the labeling scheme
Application to helical membrane proteins
Combinatorial labeling can be employed as a stand-alone technique to partially assign the protein backbone providing site-specific probes that can be useful in certain instances, as described also by (Parker et al. 2004). Whereas their method suggests half labeling of certain residues to further increase the information content, the UPLABEL introduced here solely relies on the absence or presence of a peak within the different samples. Although this requires preparation of more samples, analysis is clearly advantageous for the application to membrane proteins where high peak intensity variations occur within the sequence as for instance between loop and transmembrane regions. Conformational exchange phenomena further complicate the analysis.
As an example, the UPLABEL algorithm was applied to the bradykinin receptor B2R. This G-protein-coupled receptor is activated upon binding of the peptide hormone bradykinin whose structure has been solved in complex with its receptor (Lopez et al. 2008). Structural data on the receptor itself is sparse mostly due to limited availability of functionally reconstituted protein. To further investigate the mechanisms of peptide binding and to use these insights for drug design, data on the receptor would be highly desirable. Unique pair labeling could provide a powerful tool to assign this 44 kDa complex that comprises 391 amino acid residues and 115 unique pairs excluding prolines, either covering the complete sequence or focusing on the peptide binding pocket as demonstrated here. Fig. 3 shows the sequence of B2R with a labeling scheme determined by the UPLABEL algorithm which was limited to 9 samples. A total of 100 iterations were conducted with weighting factors in Eq. (1) of w 1 = 10, w 2 = 0, w 3 = 10, w 4 = 0 using about 7 h of computation time on a standard desktop computer. This representation allows for an easy evaluation of the distribution of labeled amino acid pairs in an amino acid sequence. For 108 out of all 115 unique pairs, i.e. those with ‘1’ in the first row and ‘X’ in the second row, the labeling patterns are unique. These pairs can be assigned unambiguously by this labeling scheme which requires the preparation of 9 samples. The number of unique pairs that can be assigned unambiguously by a labeling scheme with a given number of samples, n, is shown in Table 1. It falls short of the theoretical maximum of 2n−1 because the co-labeling of pairs precludes the use of some labeling patterns. With ten samples all unique pairs can be identified unambiguously by their labeling pattern.

Amino acid sequence, unique pairs, and labeling scheme obtained with the UPLABEL algorithm for the human G-protein coupled receptor bradykinin B2 (B2R). In the upper panel the labeling patterns of the pairs in the nine samples are shown below the sequence, indicating labeled pairs with an ‘X’ between the two amino acids that form a pair. The two rows above the sequence show, respectively, the number of occurrences of amino acid pairs (‘1’ for unique pairs, ‘2’ for pairs that occur twice in the sequence, etc.), and the unique pairs that can be assigned unambiguously (indicated by ‘X’) with the given labeling scheme. In the lower panel five different labeling schemes, each comprising nine samples, are reported side-by-side for inspection by the spectroscopist. Columns correspond to samples, and rows correspond to the 20 standard amino acid types. The labeling of each amino acid type in a given sample is indicated by ‘C’ for 13C labeling, ‘N’ for 15N labeling, and ‘B’ for 13C and 15N double labeling
Since the solution space is searched randomly, multiple runs of the algorithm using different random number generator seed values will usually produce slightly different results (Table 1). Even if two labeling schemes assign the same number of unique pairs that does not imply that both labeling schemes are the same. Many different labeling schemes exist that unambiguously assign the same number of (but not the same) unique pairs. To allow for the manual selection of a suitable labeling scheme the 5–10 best solutions are kept for inspection (Fig. 3). The user can then select one of them based on possible further criteria that are not captured by the scoring function.
In order to look at a certain region of the protein sequence, it is possible to restrict the search for assigning unique pairs. For this, it is not sufficient to simply extract the sequence regions of interest and to compute an optimal labeling scheme for this isolated part of the sequence because the rest of the sequence can also give rise to peaks. If pairs are labeled outside the selected range, they can be used to confirm existing assignments, if available, but only unique pairs within the regions of interest are to be assigned unambiguously based on the labeling scheme. To demonstrate this, we determined an optimal labeling scheme for residues 131–172 of the protein B2R. This region corresponds to part of its ligand binding site, which involves helices 3, 6, and 7, and contains 17 unique pairs. A total of 100 iterations were conducted with 4 samples, weighting factors w 1 = 10, w 2 = 0, w 3 = 10, and w 4 = 50. All amino acids types except proline were considered. Using 4 samples, this labeling scheme yields unambiguous assignments for 7 unique pairs, of which 6 are in the range of interest of residues 131–172 (Fig. 4).

Labeling scheme optimized for residues 131–172 of the human G-protein coupled receptor bradykinin B2 (B2R). Symbols have the same meaning as in Fig. 3
In many cases, however, the assignment does not solely rely on unique pair labeling, but is combined with additional data, for instance from uniformly labeled samples. In solution NMR, assignment of membrane proteins mostly relies on the most robust and sensitive experiments as the HNCA, HNCOCA for assignment while information from the HNCACB is often incomplete. Without the Cβ shift that is more characteristic for the amino acid type than the Cα shift and provides an additional sequential connectivity, assignment is often highly ambiguous. In this respect, combinatorial labeling can be helpful at two stages. At the outset of the assignment process it can serve as a method to provide starting points for the assignment. To this end the labeling is designed to cover a large percentage of the sequence by using abundant amino acids such as Ala, Gly, Ile, Leu, Phe, and Val that tend to cluster in the transmembrane regions (Reckel et al. 2008). The number of unique pairs is then less important as ambiguities can be resolved with the help of uniform labeling. In the case of the 26 kDa membrane protein proteorhodopsin a combinatorial labeling approach comprising 15N-Ala, Phe, and Ile combined with 1-13C-Ser, Leu, Val, and Gly was crucial to start the assignment. Preparation of three different samples as indicated in Table 2 allowed the differentiation between all alanines, phenylalanines and isoleucines, respectively, and enabled the assignment of 4 out of 20 alanines as part of a Ser-Ala pair, 4 others as part of a Leu-Ala pair and another 2 as part of a Val-Ala pair. Likewise, seven pairs with phenylalanine (1 Ser-Phe, 2 Leu-Phe, 4 Val-Phe), and six pairs with isoleucine (4 Leu-Ile, 2 Gly-Ile) could be identified in the combination of HSQC and 2D HN(CO) experiments (Fig. 5). Additional information about the residue types is a positive side effect although it can obviously also be obtained by standard selective labeling approaches.

Unique pair labeling of proteorhodopsin. The upper row shows the 15N,1H-TROSY-HSQC spectra (A, C, E) of three samples labeled as indicated in Table 2, with the related 2D 15N,1H HN(CO) spectra (B, D, F) below. As an example, the same peak position is encircled in each spectrum. While the peak can be observed in the 15N,1H-TROSY-HSQC spectra of samples 1 and 2, it is absent in sample 3 and can thus be identified as a phenylalanine. Further specification can then be done together with the 2D HN(CO) where the peak is only observed in sample 2 which indicated a valine as the preceding residue. Together with uniform labeling this peak was assigned as Phe76
Unique pair labeling can also be employed at a later stage of the assignment process when a significant portion of the backbone resonances have been identified using, for example, standard assignment procedures with uniformly labeled samples. Due to conformational exchange broadening, peak overlap or the presence of prolines, gaps in the assignment will occur. These regions can then be assigned using a site-specific combinatorial labeling strategy focusing on amino acids of the respective region as described above for the bradykinin receptor.
Conclusions
In this paper we introduced a new algorithm for optimizing amino acid type-specific labeling to determine backbone assignments of proteins that cannot be assigned unambiguously by other methods. The UPLABEL algorithm provides more flexibility than earlier approaches (Maslennikov et al. 2010; Parker et al. 2004; Trbovic et al. 2005). The main difference to (Parker et al. 2004) is that in UPLABEL the peak patterns are defined exclusively by the presence or absence of peaks, whereas (Parker et al. 2004) consider peak heights which can be difficult to assess in case of low signal-to-noise and overlap. The number of samples and a variety of conditions can be specified for a given sequence. This can be useful to obtain optimal results with an affordable effort and cost of sample production. The protein samples that are produced for obtaining backbone assignments with the UPLABEL algorithm can subsequently also be used to collect long-range conformational restraints for structure calculations. The advantages of extensive and selective labeling are reduced spectral overlap due to the reduction of the number of signals and a marked decrease of line-widths due to the removal of scalar and dipolar couplings, which is beneficial for the collection of conformational restraints.
References
Ausiello G, Datri A, Protasi M (1980) Structure preserving reductions among convex optimization problems. J Comput Syst Sci 21:136–153
Busche AE, Aranko AS, Talebzadeh-Farooji M, Bernhard F, Dötsch V, Iwai H (2009) Segmental isotopic labeling of a central domain in a multidomain protein by protein trans-splicing using only one robust DnaE intein. Angew Chem 48:6128–6131
Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H (2002) Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 420:98–102
Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M (2007) Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew Chem Int Edit 46:459–462
Franks WT, Wylie BJ, Schmidt HLF, Nieuwkoop AJ, Mayrhofer RM, Shah GJ, Graesser DT, Rienstra CM (2008) Dipole tensor-based atomic-resolution structure determination of a nanocrystalline protein by solid-state NMR. Proc Natl Acad Sci USA 105:4621–4626
Güntert P (1997) Calculating protein structures from NMR data. Meth Mol Biol 60:157–194
Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, van Rossum BJ, Oschkinat H (2009) Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR 44:245–260
Hong M (1999) Determination of multiple ϕ-torsion angles in proteins by selective and extensive 13C labeling and two-dimensional solid-state NMR. J Magn Reson 139:389–401
Kainosho M, Güntert P (2009) SAIL: stereo-array isotope labeling. Q Rev Biophys 42:247–300
Kainosho M, Tsuji T (1982) Assignment of the three methionyl carbonyl carbon resonances in Streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution. Biochemistry 21:6273–6279
Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Ono AM, Güntert P (2006) Optimal isotope labelling for NMR protein structure determinations. Nature 440:52–57
Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C (2008) The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy. Angew Chem Int Edit 47:1668–1671
Lundström P, Teilum K, Carstensen T, Bezsonova I, Wiesner S, Hansen DF, Religa TL, Akke M, Kay LE (2007) Fractional 13C enrichment of isolated carbons using [1–13C]- or [2–13C]-glucose facilitates the accurate measurement of dynamics at backbone Cα and side-chain methyl positions in proteins. J Biomol NMR 38:199–212
Maslennikov I, Klammt C, Hwang E, Kefala G, Okamura M, Esquivies L, Mörs K, Glaubitz C, Kwiatkowski W, Jeon YH, Choe S (2010) Membrane domain structures of three classes of histidine kinase receptors by cell-free expression and rapid NMR analysis. Proc Natl Acad Sci USA
McCaldon P, Argos P (1988) Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins 4:99–122
Parker MJ, Aulton-Jones M, Hounslow AM, Craven CJ (2004) A combinatorial selective labeling method for the assignment of backbone amide NMR resonances. J Am Chem Soc 126:5020–5021
Reckel S, Sobhanifar S, Schneider B, Junge F, Schwarz D, Durst F, Löhr F, Güntert P, Bernhard F, Dötsch V (2008) Transmembrane segment enhanced labeling as a tool for the backbone assignment of α-helical membrane proteins. Proc Natl Acad Sci USA 105:8262–8267
Schmucki R (2008) Peak particle dynamics for automated NMR resonance assignment. Ph.D. thesis, The University of Tokyo, Tokyo
Schmucki R, Yokoyama S, Güntert P (2009) Automated assignment of NMR chemical shifts using peak-particle dynamics simulation with the DYNASSIGN algorithm. J Biomol NMR 43:97–109
Schneider R, Ader C, Lange A, Giller K, Hornig S, Pongs O, Becker S, Baldus M (2008) Solid-state NMR spectroscopy applied to a chimeric potassium channel in lipid bilayers. J Am Chem Soc 130:7427–7435
Schubert M, Manolikas T, Rogowski M, Meier BH (2006) Solid-state NMR spectroscopy of 10% 13C labeled ubiquitin: spectral simplification and stereospecific assignment of isopropyl groups. J Biomol NMR 35:167–173
Shi J, Pelton JG, Cho HS, Wemmer DE (2004) Protein signal assignments using specific labeling and cell-free synthesis. J Biomol NMR 28:235–247
Skrisovska L, Allain FHT (2008) Improved segmental isotope labeling methods for the NMR study of multidomain or large proteins: Application to the RRMs of Npl3p and hnRNP L. J Mol Biol 375:151–164
Trbovic N, Klammt C, Koglin A, Löhr F, Bernhard F, Dötsch V (2005) Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc 127:13504–13505
van Gammeren AJ, Hulsbergen FB, Hollander JG, de Groot HJM (2004) Biosynthetic site-specific 13C labeling of the light-harvesting 2 protein complex: A model for solid state NMR structure determination of transmembrane proteins. J Biomol NMR 30:267–274
Yamazaki T, Otomo T, Oda N, Kyogoku Y, Uegaki K, Ito N, Ishino Y, Nakamura H (1998) Segmental isotope labeling for protein NMR using peptide splicing. J Am Chem Soc 120:5591–5592
Zech SG, Wand AJ, McDermott AE (2005) Protein structure determination by high-resolution solid-state NMR spectroscopy: Application to microcrystalline ubiquitin. J Am Chem Soc 127:8618–8626
Zhou DH, Shea JJ, Nieuwkoop AJ, Franks WT, Wylie BJ, Mullen C, Sandoz D, Rienstra CM (2007) Solid-state protein-structure determination with proton-detected triple-resonance 3D magic-angle-spinning NMR spectroscopy. Angew Chem Int Edit 46:8380–8383
Züger S, Iwai H (2005) Intein-based biosynthetic incorporation of unlabeled protein tags into isotopically labeled proteins for NMR studies. Nat Biotechnol 23:736–740
Acknowledgments
We gratefully acknowledge financial support by the Lichtenberg program of the Volkswagen Foundation and by a Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (JSPS). S. J. U. is supported by the Studienstiftung des deutschen Volkes.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hefke, F., Bagaria, A., Reckel, S. et al. Optimization of amino acid type-specific 13C and 15N labeling for the backbone assignment of membrane proteins by solution- and solid-state NMR with the UPLABEL algorithm. J Biomol NMR 49, 75–84 (2011). https://doi.org/10.1007/s10858-010-9462-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-010-9462-4