
Abstract
This work investigated the effects of CeO2 contents on structure, crystallization behavior and dielectric properties of CaO–B2O3–SiO2 glass composition. The MAS-NMR results showed that B occurred as BIIIa, BIIIs and BIV species and Si presented as Q2, Q3 and Q4 units in the glasses. As the increase of CeO2 content, the relative amounts of BIV and BIIIs unit decreased while the BIIIa units increased, and the amounts of Q4 and Q2 units increased while the Q3 unit decreased. With increasing CeO2 content, the value of Tg decreased from 743 °C to 717 °C, the activation energy for CaSiO3 first increased and then decreased. For glass–ceramics samples sintered at 825 °C, all samples had the crystalline phases of CaSiO3, CaB2O4, Ca2SiO4. In addition, the samples with CeO2 content more than 1 mol% had CeO1.695 phase, which changed to be the main crystalline phase when the content of CeO2 increased up to 10 mol%. The εr of the glass–ceramic samples with CeO2 content x ≤ 6 showed an ascend trend in total, and decreased sharply to 4.1 for the sample with x = 10. However, the dielectric loss tan δ would not change significantly with the increasing of CeO2 content. The samples with 1 mol% CeO2 sintered at 825 °C owned εr of 5.4, tan δ of 0.9 × 10− 3, and CTE of 7.8 × 10− 6/K. The results indicated that CaO–B2O3–SiO2 with CeO2 glass–ceramics could be a potential LTCC substrate material.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, miniaturization and integration development of electronic components have set a higher demand for the substrates, which should satisfy the high propagation speed, high wiring density and large chip packaging [1, 2]. The substrate materials of low temperature co-fired ceramic (LTCC) technology should possess low dielectric constant and loss, low coefficient of thermal expansion (CTE) to match with silicon (3 × 10− 6/K) and low sintering temperature (< 900 °C) for co-firing with metal electrodes such as Cu, Ag and Au [2, 3]. Among the multiple glass systems utilized in LTCC technology, CaO–B2O3–SiO2 glasses take up a crucial position due to its complete sintering properties at temperatures below 900 °C with low dielectric constant (εr < 6.5) and dielectric loss (tan δ < 1 × 10− 3) [4, 5] along the last decades.
However, CaO–B2O3–SiO2 glasses also exhibit some unfavorable characteristics for the development of future high frequency communication. Their relatively low mechanical strength, narrow sintering window and dielectric properties will hinder their sufficient use in LTCC technological area. In order to satisfy a high level of need of the technical applications, the optimization in preparation method, composition and structure of CaO–B2O3–SiO2 glasses is carried out to achieve the required properties [6, 7]. Xia et al. [8] developed a high-performance film for LTCC application through using a kind of silane coupling agent (SCA) to modify the CaO–B2O3–SiO2 glass–ceramic powders in tape casting process. The experimental results revealed that the powders modified by SCA with 1.5 wt% additions owned the optimal performance of film, possessing the highest density and best microwave properties of sintered body: the density of sintered body was 2.48 g/cm3, the dielectric constant and dielectric loss were εr = 5.93, tan δ = 8 × 10− 4 at 12 Hz. Lee et al. [9] investigated carbon nanotube/graphene oxide-added CaO–B2O3–SiO2 glass/Al2O3 composite. The results showed that the CaO–B2O3–SiO2 glass/40 wt% Al2O3 composite containing 0.25 wt% graphene oxide and 0.75 wt% carbon nanotube, sintered at 900 °C and subsequently annealed at 850 °C, exhibited a high bending strength of 420 MPa and thermal conductivity of 5.51 W/(mK). These outstanding properties make this composite suitable as a substrate for chip-type supercapacitors.
Moreover, some oxides, such as ZnO [10, 11], MgO [12], ZrO2 [13] and Al2O3 [14, 15], were introduced into CaO–B2O3–SiO2 glass to modify the structure of glass to achieve the required physical, mechanical and dielectric properties. Park et al. [16] studied 50CaO·20ZnO·20B2O3·10SiO2 glass, which had CTE of 12.8 × 10− 6/K and dielectric constant of 13.5. The results indicated the feasibility of the developed glass for the application to LTCC materials with high CTE. Liu et al. [15] prepared CaO–B2O3–SiO2 glass ceramics sample with 5 wt% Al2O3, possessing the best sintering characterization and dielectric properties (εr ≈ 7, tan δ = 1.9 × 10− 3 at 1 MHz), which was proposed to be suitable for LTCC application. According to our previous report [17], the CaO–B2O3–SiO2 glasses with addition of 5 wt% La2O3 had the best dielectric properties (εr ≈ 4.1, tan δ ≈ 1.7 × 10− 3 at 15 GHz), which exhibited the great potential of rare-earth doping to improve the properties of CaO–B2O3–SiO2 systems. In addition, it was interesting that the addition of CeO2 could significantly change the crystallization behavior and microwave dielectric properties of glass–ceramic [18,19,20]. Liu et al. [20] reported the effect of CeO2 on the SrO–BaO–Nb2O5–B2O3–SiO2 glass–ceramics, which showed the addition of CeO2 decreased the activation energy of crystallization and enhanced the dielectric constant as well as breakdown strength remarkably.
However, the effects of CeO2 on crystallization behavior and dielectric properties of CaO–B2O3–SiO2 glass system have not been taken seriously. In this work, we systematically investigated the effects of CeO2 on glass structure, crystallization behavior, sintering behavior and dielectric properties of CaO–B2O3–SiO2 glasses, expecting to develop an optimized material with outstanding dielectric properties for further LTCC application.
2 Experimental
2.1 Preparation of glass powders
The (100 − x)[40CaO–37SiO2–23H3BO3] + xCeO2 glasses, where x = 0, 0.5, 1, 3, 6 and 10 mol% (noted as C0, C0.5, C1, C3, C6, and C10, respectively) were prepared using AR grade CaCO3, H3BO3, SiO2, and CeO2 as the starting materials. The glass powders were prepared via a water quenching method as reported in our previous work [14].
2.2 Preparation of glass–ceramic samples
The glass powder was granulated with a 4 wt% polyvinyl alcohol (PVA) solution and compacted into disk-shaped specimens under the uniaxial pressure of about 100 MPa. Then the samples were sintered for 20 min in air at temperature ranging from 750 °C to 850 °C with a heating rate of 5°C/min.
2.3 Characterization of the samples
The X-ray diffraction XRD (XRD, DMX-2200, Rigaku, Tokyo, JP) of glass powders and glass–ceramic samples was measured using a standard diffractometer with Cu Kα radiation (λ = 1.54 Å) as an X-ray source. All samples were scanned from 10° to 80° at step width of 0.02°. 11B and 29Si magic angle spinning nuclear magnetic resonance spectroscopy (MAS-NMR, Varian Infinity-plus 400, US) were applied to analyze the structure of B and Si units in glass system. The samples C0, C0.5, C3 and C10 were selected to display the structure change of B and Si with increasing of CeO2 content. The measure condition refers to a reference [21].
The Fourier transform infrared spectra (FT-IR) of the glasses were recorded at room temperature using the KBr technique by a Thermo Nicolet Smart-380 FT-IR spectrometer in a wave number region between 400 and 2000 cm−1 with a resolution of 2 cm−1.
To determine the glass transition temperature and the crystallization kinetics of the glasses, a differential scanning calorimetry (DSC, STA409 PG/PC, Netzsch, Germany) were carried out in air from room temperature to 1000 °C with heating rates of 5, 10, 15 and 20 °C/min with a reference material of α-alumina powders.
The densities of glass–ceramic samples were measured by the Archimedes method. The surface morphologies of glass–ceramic samples were studied using a field emission scanning electron microscope (Ultra 55, ZEISS). The surface of samples was eroded 60 s using 2% HF solution. The network analyzer (N5230C, Agilent) was utilized to measure the dielectric properties of all samples at 15 GHz. The coefficient of thermal expansion (CTE) of all samples was carried out in argon atmosphere from 25 to 500 °C with heating rates of 5°C/min by a thermal expansion analyzer (DL 402, Netzsch).
3 Results and discussion
The XRD patterns of all glass samples are presented in Fig. 1. According to the experimental results, the diffraction peak curves, without crystalline peaks, indicated the amorphous state and good glass forming ability of all samples.

X-ray diffraction patterns of all glass samples
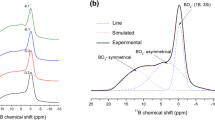
The 11B MAS-NMR spectra of C0, C0.5, C3 and C10 samples are shown in Fig. 2a. For sample C0, it is worth noting that two relatively broad peaks centered at 4.3 ppm and 10.5 ppm, which are associated with asymmetric boron units (BIIIa, boron connected with 1 or 2 bridging oxygens) and symmetric trigonal boron units (BIIIs, boron connected with 0 or 3 bridging oxygens) [14, 22, 23], respectively. The sharper peak located at − 0.2 ppm could be assigned to tetrahedral (BO4, BIV) boron species, which is danburite like structural units of boron where each tetrahedral boron is coordinated with one boron atom and three Si tetrahedrons [B(OB)(OSi)3] [14, 23]. By performing CeO2 addition, the peak shifts to higher values of the chemical shifts i.e. from − 0.2 to 3.1 ppm for x = 0 and x = 10, respectively. In order to quantify the relative amounts of BIIIa, BIIIs and BIV units of boron in glasses, the 11B MAS-NMR spectra were deconvoluted and an example of C0 was presented in Fig. 2b. The NMR parameters, the isotropic chemical shift (δiso) and the amount of boron structure units were listed in Table 1. The relative amount of BIV units decreases from 29% to 8% and BIIIs is from 26% to 20% then disappears, while the amount of BIIIa units increases from 45% to 92% with the increasing of CeO2 content (Table 1).

11B MAS-NMR spectra of C0, C0.5, C3 and C10 (a) and deconvolution spectra of C0 (b)
Figure 3a reveals the 29Si MAS-NMR spectra of C0, C0.5, C3 and C10 samples. It can be noted that for all glass samples, the spectra present a peak maximum near − 84 ppm. Deconvolution of 29Si spectra was performed using Gaussian to quantitatively clarify the fractions and distribution of different Si units in glasses. An example of C0 was presented in Fig. 3b, the isotropic chemical shifts (δiso) and the relative amounts of different Si units were listed in Table 1. The peaks for all glasses around − 82 ppm, − 87 ppm and − 109 ppm, which are characteristic of the Q2, Q3 and Q4 structural units of silicon, respectively [24]. With the increasing of CeO2, the amount of Q4 unit increases from 0% to 36% and the Q2 increases from 25% to 50%, while the amount of Q3 unit decreases from 75% to 14%.

29Si MAS-NMR spectra of C0, C0.5, C3 and C10 (a) and deconvolution spectra of C0 (b)
The MAS-NMR results had shown that B occurred as BIIIa, BIIIs and BIV (1B,3Si) species and Si presented as Q2, Q3 and Q4 units in the glasses. As the increase of CeO2 content, the relative amount of BIV and BIIIs units decreased while the amount of BIIIa units increased, and the amount of Q4 and Q2 unit increased while the amount of Q3 units decreased. The change of boron species could be ascribed to the CeO2 in glass system. Generally, the six or eight coordination numbers and ionic field strength of 0.83 [25] make cerium play the part of network modifier in the glass system, and it prefers to bridge BO3 units rather than form BO4 groups [26]. This will make the amount of BO3 units increase. Moreover, cerium oxide is considered to form Ce–O–B(3) bonds at the expense of the bridging B(3)–O–B(3) bonds [26], which leads to the increase of BIIIa and the decrease of BIIIs. Additionally, according to our previous study [14], the BIV species can resolve into symmetric BO3 species and non-bridging oxygen (NBO), the BIIIs species with the NBO can form BIIIa species, both of which resulting in the change of boron structural units. On the other hand, the non-bridging oxygen created by BIV resolving and [CeO6] or [CeO8] unit forming [20] in the glass may react with Qn to form a Qn−1 species. The structure of Q4 unit is most stable among all [SiO4] tetrahedrons so that it is difficult to react with NBO to form Q3 unit [17]. But the existing Q3 unit will react with NBO to generate Q2 unit, which make the amount of Q2 unit increase. Furthermore, the added CeO2 in glass will release a lot of free oxygen, which will occur in the formation of Si–O link to cause sectional Q3 unit turning into Q4 unit. Therefore, the amount of Q4 and Q2 unit increased while the amount of Q3 units decreased with the increasing of CeO2 content.
Figure 4 displays the FT-IR spectra between 400 and 2000 cm−1 for of all glasses. As it shown, the broad bands confirmed the amorphous nature (as shown in Fig. 1) and wide distribution of Qn units in glass systems. Seven absorption bands were observed in all glasses. Their peak positions and intensity are related to the CeO2 contents obviously. The assignments are shown in Table 2. The band located at 1427 cm−1 shifted to lower wavenumber obviously with the increasing of CeO2 content, the wavenumber locations were separately 1427, 1412, 1407, 1404, 1402, 1400 cm−1 from x = 0 to x = 10, respectively. In addition, the peak located at 1041 cm−1 became sharper with the increase of CeO2. In addition, new band didn’t appear with the increasing of CeO2 content, indicated that the CeO2 may only play the part of network modifier in CaO–B2O3–SiO2 glasses.

The FT-IR spectra of the glasses
Figure 5a presents the DSC plots of all the glasses measured at β = 5°C/min. For all samples, the curves showed 2 crystallization peaks (TP1 and TP2), related to the formation of the crystalline phase of wollastonite [32] and borocalcite [33], respectively. The values of glass transition temperature Tg, which obtained from the onset of slope change of the DSC curves, and TP1, TP2 for all glasses were listed in Table 3. With increasing CeO2 content, the value of Tg decreased from 743 °C to 717 °C. This could be ascribed to the free oxygen, which brought by CeO2 addition, destroyed the glass network structure and impaired the glass network polymerization degree. The exothermic peak temperatures TP1 of glasses first increased and then decreased obviously with the increasing of CeO2 content. It meant that a little bit of CeO2 addition could make CaSiO3 crystallization difficult, but this system would be easier to crystallize when the addition of CeO2 content exceeded 3 mol%. In addition, the TP2 increased obviously with the increasing of CeO2 content, which showed that the CeO2 inhibited the crystallization of CaB2O4. The DSC curves at β = 10, 15, 20 °C/min were also measured (the crystallization exothermal peak temperatures are listed in Table 3) in order to analyze the crystallization kinetics of glasses, which can be described by the expression [20]:

DSC curve of the studied glasses at β = 5°C/min (a) and plots of ln (β/Tp12) versus 1/Tp1 (b)
where Tp is the crystallization peak temperature in a DSC curve, β is the heating rate of DSC, R is the gas constant, and E is the activation energy for crystallization. According to the equation, a plot of (β/Tp12) versus 1/Tp1 yields a straight line in which the slopes are proportional to the activation energy of crystallization. These plots are shown in Fig. 5b and the values of crystallization activation energy for CaSiO3 are shown in Table 3. It was found that the activation energy first increased and then decreased with the increasing CeO2 content, which conformed to the change rule of TP1. The change of crystallization activation energy may be attributed to the Qn variation in glass structure.
Figure 6a–c show the XRD patterns of all glass–ceramics samples sintered at 750 °C, 775 °C and 825 °C for 20 min, respectively. For samples sintered at 750 °C (as shown in Fig. 6a), it could be observed that C0 sample remained the glassy state and samples with CeO2 content occur a slight crystallization phenomenon. The samples C0.5, C1 and C3 only had a bit of CaSiO3 phase (PDF standard card: 00-003-0626), while the CeO1.695 phase (PDF standard card: 01-089-8429) started to appear in samples C6 and C10. For glass–ceramics samples sintered at 775 °C (as shown in Fig. 6b), all samples had CaSiO3 phase. With the increase of CeO2 content, the intensity of CeO1.695 diffraction peak gradually increased, which was accompanied by a decrease in CaSiO3. When the content of CeO2 increased to 10 mol%, the main crystalline phase changed to be CeO1.695. For glass–ceramics samples sintered at 825 °C (as shown in Fig. 6c), all samples had crystalline phases of CaSiO3, CaB2O4 (PDF standard card: 00-001-0833), Ca2SiO4 (PDF standard card: 01-087-1259). In addition, the samples with CeO2 content more than 1 mol% had CeO1.695 phase, which changed to be the main crystalline phase when the content of CeO2 increased to 10 mol%. Figure 6d shows the XRD patterns of sample with 1 mol% CeO2 addition sintered at different temperatures. The sample sintered at 750 °C was amorphous. After calcination of glass–ceramic at 775 °C, CaSiO3 phase as the main phase was observed. When the temperature was above 800 °C, a couple of minor XRD peaks for CaB2O4 were also detected. The XRD patterns of the glass–ceramics samples sintered at 800 °C and 850 °C (not shown here) had the similar change rule with samples sintered at 775 °C and 825 °C respectively, indicating that all samples will gradually crystallize with the increasing of sintering temperature and achieve the complete crystallization at 825 °C.

X-ray diffraction patterns of glass–ceramics samples sintered at a 750 °C, b 775 °C, c 825 °C and d sample with 1 mol% CeO2 addition sintered at different temperatures
The values of the bulk density of the glass–ceramics samples sintered at different temperatures are depicted in Fig. 7. The densities of all samples first decreased with the increasing sintering temperature and then remained about the same when sintering temperature was above 800 °C. This is because all the samples have been completely crystallization and reach a steady state when the sintering temperature is up to 825 °C. The results are in accordance with previous outcome revealed in XRD. With the increase of CeO2 content, the densities of the samples (Table 3) sintered at 825 °C first increased slowly from 2.4 g/cm3 to 2.7 g/cm3 when the CeO2 content increase from 0 to 6 mol%, and then dropped dramatically to 1.8 g/cm3 when the content of CeO2 increased to 10 mol%. Considering the results of XRD and densities, the optimum sintering temperature of glass–ceramics samples was 825 °C.

Densities of glass–ceramics samples sintered at different temperatures
Figure 8 displays the SEM micrographs of the surface of the glass–ceramics samples sintered at 825 °C for 20 min with different CeO2 contents. It was observed that the all glass–ceramics samples were mainly composed of the crystal phase (spherical particles) and the glass phase. For C0 to C3 samples, the amounts of spherical particles, which mainly corresponded to CaSiO3 phase (as shown in Fig. 6c), decreased obviously. This result is consistent with DSC data where the values of the activation energy increase from C0 to C3 gradually. In addition, the uniform and dense microstructure with grain sizes around 500 nm could be observed in samples C0, C0.5 and C1. With the further increase of CeO2 contents, CeO2 deteriorated the uniform microstructure (as shown in C3), which had the smallest grain size approaching 100 nm and the largest grain size ~ 700 nm. For C6 sample, the grain size greatly decreased to < 100 nm and the amount of grains increased significantly. Upon the addition of 10 mol% CeO2, the grain size kept growing and had the average grain size about 200 nm, with the smallest grain size < 100 nm and the largest grain size ~ 400 µm. Based on the results of XRD (Fig. 6c), the grains in C6 and C10 mainly are CeO1.695 phase.

SEM photographs of the surface of the glass–ceramics sintered at 825 °C with different CeO2 contents
The microwave dielectric properties of the glass–ceramics sintered at 825 °C as a function of the CeO2 content are presented in Fig. 9. It was noticed that εr of the glass–ceramic samples with CeO2 content x ≤ 6 showed an ascend trend in total, and decreased sharply to 4.1 for the sample with x = 10, probably owing to its low density. For sample C10, the excess Ce4+, as the network modifier in the glass network, made the massive formation of CeO1.695 phase in the glass systems. This would greatly deteriorate the dielectric properties. However, the dielectric loss tan δ will not change significantly with the increasing of CeO2 content, within a small scope of 0.9–1.2 × 10− 3. The results reveal that the dielectric properties of the glass–ceramics are closely related to density and microstructure. In addition, the coefficients of thermal expansion (CTE) averaging from 25 °C to 500 °C for glass–ceramic samples sintered at 825 °C are listed in Table 3. The CTE of the glass–ceramic samples with CeO2 content had slightly difference compared to the value 7.6 × 10− 6/K of C0.

The microwave dielectric properties of glass–ceramic samples sintered at 825 °C as a function of the CeO2 content
4 Conclusions
In this work, we investigated the structure, crystallization behavior and dielectric properties of CaO–B2O3–SiO2 glass with CeO2 addition. The NMR results revealed that the Ce played the network modifier role in CaO–B2O3–SiO2 glasses, and B went into the glass network as both fourfold and threefold coordination whereas the Si occurred in the form Q2, Q3 and Q4 units. With the increasing of CeO2 content, the relative amount of BIV and BIIIs units decreased while the amount of BIIIa units increased, and the amount of Q4 and Q2 unit increased while the amount of Q3 units decreased. The addition of CeO2 has been found to decrease Tg from 743 °C to 717 °C. For the glass–ceramics samples sintered at 825 °C, the crystalline phases of C0 were CaSiO3, CaB2O4, Ca2SiO4. Upon the addition of CeO2 more than 1 mol%, the CeO1.695 phase occurred and changed to be the main crystalline phase when the content of CeO2 increased to 10 mol%. After the analysis and comparison, we could conclude that the C1 sample had the optimum properties (εr: 5.4, tan δ: 0.9 × 10− 3, CTE: 7.8 × 10− 6/K), which indicated the potential application for LTCC substrate.
References
Z. Qing, Mater. Lett. 212, 126 (2018)
K. Manu, M.T. Sebastian, Ceram. Int. 42, 1210 (2016)
X. Zhou, E. Li, S. Yang, B. Li, B. Tang, Y. Yuan, S. Zhang, Ceram. Int. 38, 5551 (2012)
H. Zhu, H. Zhou, M. Liu, P. Wei, G. Xu, G. Ning, J. Mater. Sci. Mater. Electron. 20, 1135 (2008)
C.C. Chiang, S.F. Wang, Y.R. Wang, Y.F. Hsu, J. Alloys Compd. 461, 612 (2008)
Y. Lai, Y. Zeng, X. Tang, H. Zhang, J. Han, H. Su, RSC Adv. 6, 93722 (2016)
S.F. Wang, Y.R. Wang, Y.F. Hsu, C.C. Chiang, J. Alloys Compd. 498, 211 (2010)
Y. Xia, Y. Hu, L. Ren, X. Luo, W. Gong, H. Zhou, J. Eur. Ceram. Soc. 38, 253 (2018)
T.H. Lee, S.H. Cho, T.G. Lee, H.T. Kim, I.K. You, S. Nahm, J. Am. Ceram. Soc. 101, 3156 (2018)
J. Han, Y. Lai, Y. Xiang, S. Wu, Y. Xu, Y. Zeng, J. Chen, J. Liu, J. Mater. Sci. Mater. Electron. 28, 6131 (2017)
S. Cetinkaya Colak, I. Akyuz, F. Atay, J. Non-Cryst. Solids 432, 406 (2016)
H. Shao, T. Wang, Q. Zhang, J. Alloys Compd. 484, 2 (2009)
S. Khan, G. Kaur, K. Singh, Ceram. Int. 43, 722 (2017)
J. Han, Y. Lai, Y. Xiang, S. Wu, Y. Zeng, H. Yang, Y. Mao, Y. Yang, RSC Adv. 7, 14709 (2017)
J.Z. Liu, X.F. Wu, N.X. Xu, Q.L. Zhang, H. Yang, J. Mater. Sci. Mater. Electron. 26, 8899 (2015)
J.S. Park, Y. Kim, H. Shin, J.H. Moon, W. Lim, J. Am. Ceram. Soc. 91, 3630 (2008)
Y. Xiang, J. Han, Y. Lai, S. Li, S. Wu, Y. Xu, Y. Zeng, L. Zhou, Z. Huang, J. Mater. Sci. Mater. Electron. 28, 9911 (2017)
J. Wang, W. Chen, L. Luo, J. Alloys Compd. 464, 440 (2008)
S.B. Sohn, S.Y. Choi, J. Non-Cryst. Solids 282, 221 (2001)
T.Y. Liu, G.H. Chen, J. Song, C.I. Yuan, Ceram. Int. 39, 5553 (2013)
S. Sen, Z. Xu, J.F. Stebbins, J. Non-Cryst. Solids 226, 29 (1998)
H.R. Gaddam, J.M.F. Fernandes, Ferreira, RSC Adv. 5, 41066 (2015)
G. Parkinson, D. Holland, M.E. Smith, A.P. Howes, C.R. Scales, J. Phys. Condens. Matter 19, 415114 (2007)
T. Schaller, J.F. Stebbins, M.C. Wilding, J. Non-Cryst. Solids 243, 146 (1999)
M.B. Volf, Chemical Approach to Glass (Elsevier, Amsterdam, 1984)
G. El-Damrawi, K. El-Egili, Physica B 299, 180 (2001)
G.J. Mohini, N. Krishnamacharyulu, G. Sahaya Baskaran, P.V. Rao, N. Veeraiah, Appl. Surf. Sci. 287, 46 (2013)
R.C. Lucacel, T. Radu, A.S. Tătar, I. Lupan, O. Ponta, V. Simon, J. Non-Cryst. Solids. 404, 98 (2014)
K. Singh, I. Bala, V. Kumar, Ceram. Int. 35, 3401 (2009)
M. Sitarz, J. Non-Cryst. Solids 357, 1603 (2011)
D. Winterstein-Beckmann, D. Möncke, E.I. Palles, L. Kamitsos, Wondraczek, J. Non-Cryst. Solids 405, 196 (2014)
J.H. Jean, C.R. Chang, C.D. Lei, J. Am. Ceram. Soc. 87, 1244 (2004)
C.R. Chang, J.H. Jean, J. Am. Ceram. Soc. 82, 1725 (1999)
Acknowledgements
This work was supported by the fund of the Basic Applied Research Foundation of Yunnan Province (Grant No. 2016FD125, 2016FB083), Key New Product Project of Yunnan Province (Grant No. 2016BA009), Chinese academy of Sciences Key Project (Grant No. KLIFMD201605) and 2017 Kunming Advanced Talent Funding (13020163).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Han, J., Xiang, Y., Yao, Z. et al. Glass structure, crystallization kinetics and dielectric properties of CeO2-added CaO–B2O3–SiO2 glass system. J Mater Sci: Mater Electron 30, 5902–5910 (2019). https://doi.org/10.1007/s10854-019-00888-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-019-00888-y