
Abstract
In the present work, zinc oxide (ZnO) nanorods (NRs) were prepared by a simple Microwave-assisted hydrothermal synthesis route by using a 100 ml Teflon container. The structural, crystalline nature and purity of the synthesized material was studied by powder X-ray diffraction studies. The optical absorption wavelength observed at 368 nm has been shifted to 377 nm during hydrothermal synthesis of ZnO NRs. The shifting of wavelength reduces the energy band gap from bulk state of 3.52–3.32 eV during the Microwave-assisted hydrothermal process. There is no shift in peak observed in photoluminescence spectrum between ZnO NRs and ZnO nanoparticles (NPs) but increase in temperature increases the Deep Level Emission (DLE) intensity due to the loss of oxygen during the hydrothermal process. The FTIR analysis confirms the presence of various functional groups in the ZnO NRs and NPs. The elemental, shape and surface morphology of the ZnO NRs and NPs were determined by using EDAX and FE–SEM analysis. The surface properties of ZnO NPs and ZnO NRs were studied by BET analysis. Thermal stability of the material was increased from 180 to 277 °C after the hydrothermal synthesis. The zeta potential analysis reveals the stability of the synthesized ZnO nanostructures. The variation of dielectric constant, dielectric loss and ac conductivity with frequency for the various temperatures was measured and discussed the suitability of the material for the electronic device fabrication.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent trends, one-dimensional (1-D) nanostructures have been mainly focused on fabricating and correlating the morphologies of their size-related optical and electrical properties [1,2,3,4,5]. 1-D semiconductor nanostructures, such as nanorods, nanowires and nanobelts have recently attracted much attention for their potential use as fundamental building blocks for the new generation of electronic and photonic devices [6]. Also, 1-D nanostructures reports about the various structures including hollow microspheres, tower, dumbbell, nut, flower, nanodots, nanotubes, nanobridges, nanonails, nanowalls, nanohelixes, seamless nanorings, mesoporous single-crystal nanowires and polyhedral cages like nanostructures [7,8,9,10,11,12,13,14]. In current trends, various 1-D semiconductor nanostructures such as TiO2, SnO2, GaN, GaAs, Si and ZnO have been synthesized and reported by various researchers from different part of the country [15, 16]. Among them, ZnO nanostructure has become particularly interesting because of its wide band gap energy (3.37 eV) with high excitonic binding energy (~ 60 meV) [17], enhanced semiconducting property and its wide range of applications in optoelectronics, solar cells, gas sensors and photocatalysts due to its environmental photochemistry property [18]. In general, several well-established synthesis methods involved such as sol–gel, spray pyrolysis, sputtering, hydrothermal and solvothermal were used so far in the synthesis of ZnO nanostructures [19]. Among the various methods, hydrothermal method has more advantages compare than the other synthesis techniques, which are: low aggregation level, narrow crystallite size distribution and one-step synthesis method. In addition, high purity of the materials and excellent morphology and size of the particles was controlled by this technique [20]. However, solution methods are more suitable for inexpensive mass production but the hydrothermal technique requires specialized equipment and a variable pressure gradient control which makes it a little expensive over the simple solution method and it is time intensive. In recent trends, the hydrothermal Microwave-assisted synthesis method has more attraction and wide interests as a novel heating model in materials science due to its various advantages including normal atmospheric pressure reacting, short reaction time, rapid heating, low reaction temperature, homogeneous thermal transmission and the phase purity with better yield [19]. Also, the nanostructures can easily form in this method compared to other synthesis method. Generally, in hydrothermal processes surfactants or complexing agents such as hexamethylenetetramine (HMT), cetyltrimethylammonium bromide (CTAB), citrate potassium, ethylenediamine (En) or citric acid were used to control the morphology of the materials [21,22,23,24,25,26,27,28,29,30]. In the present Microwave-assisted hydrothermal synthesis method, hydrazine hydrate (N2H4·H2O) instead of NaOH was used as mineralizer without addition of any surfactants or complexing agents. Normally, hydrazine hydrate is a very cheap and powerful reducing agent but Zhu et al., has proposed that the hydrazine hydrate forms a complex when react with the ZnCl2 solution and the complex slowly decomposed during the hydrothermal process to form ZnO NRs [31]. Meng et al. was reported Microwave-assisted hydrothermal method possesses and combines the merits of Microwave and hydrothermal methods which can achieve the high temperature and high pressure for a short time from several minutes to several hours in a closed reaction system. Also briefly described the various structures of the metal oxides and hydroxides via Microwave-assisted hydrothermal synthesized material such as ZrO2, CuO, ZnO, CeO2, TiO2, MnO2, Wo3, SnO2, MgO, α-Fe2O3 etc. used for optical, gas sensor, catalytic, solar cells and electrochemical applications [32]. Ghosh et al., was reported a comparison of conventional hydrothermal and Microwave-assisted hydrothermal process, where Microwave-assisted hydrothermal process takes less than 4 h whereas conventional hydrothermal synthesis takes 25 h to fabricate nanoforest [33]. In the present investigation, the ZnO NRs were obtained within 45 min duration. The as-prepared material was subjected to analysis the structural, morphology, optical and thermal behavior before (NPs) and after the Microwave-assisted hydrothermal treatment (NRs). A comparative analysis was made and reported for the first time. The possible nucleation and reaction mechanisms of the ZnO nanostructures were discussed. The dislocation density (δ), internal stress (ε) and strain (σ) of the ZnO NRs and ZnO NPs were also analyzed.
2 Materials and methods
2.1 Materials for synthesis of ZnO nanorods
The materials which were used for the synthesis ZnO are zinc chloride (ZnCl2) and hydrazine hydrate (N2H4·H2O). All the chemicals used in the experiment were of analytical grade and were purchased from Merck. Ultra pure water of resistivity 18.2 MΩcm from Milli-Q system (Millipore) was used for the preparation of solutions throughout the study.
2.2 Synthesis procedure for ZnO NPs and ZnO NRs
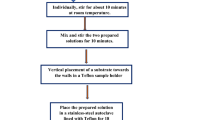
ZnO NRs were synthesized using ZnCl2 as a precursor material. Figure 1 shows the schematic synthesis procedure of the ZnO NRs. 0.54 g of ZnCl2 and 0.4 ml of N2H4 were dissolved in 50 mL of Millipore water under constant stirring. When the reaction completes white precipitate with solution is formed which is centrifuged for 20 min and dried in hot air oven at 60 °C to obtain ZnO NPs. The obtained ZnO NPs were used for further characterization.

Schematic synthesis procedure for ZnO NRs
0.54 g of ZnCl2 and 0.4 ml of N2H4 were dissolved in 50 mL of Millipore water under constant stirring. When the reaction completes white precipitate was formed and that solution was transferred to a Teflon container. The mixture was radiated (400 W, 100%) in 45 min at 160 °C in Microwave oven type IFB 17PG2S (frequency 2.45 GHz, maximum power 1200 W) and then cooled at room temperature naturally. Pure white precipitate was formed at the bottom of the container. The product was centrifuged twice and rinsed with alcohol and deionized water for several times and dried at 80 °C in air. The schematic of the synthesis procedure for ZnO NRs is shown in Fig. 1.
2.3 Reaction mechanism
The chemical mechanism for the formation of ZnO NP and ZnO NR in before and after hydrothermal can be explained by the following equations:
The morphologies of the nanomaterials were depends upon the nucleation and growth of the crystal direction [34]. Initially, before and after hydrothermal process, the slurry like precursor (ZnCl2(N2H4) n ) was formed via reaction (1). In the reaction (2), the ZnCl2(N2H4) n complex underwent dissociation and hence independent Zn2+ and N2H4 were formed. Then N2H4 start to decompose slowly and hence produces NH3 in the reaction (3) and during the reaction (4) Zn(OH)2 anions were formed before and after hydrothermal process because hydroxide (Zn(OH)2) anions are formed very quickly due to its strong basicity (or) strong reducibility of N2H4 [35]. In before hydrothermal, Zn(OH)2 gives Zn2+ and OH− which gives super saturation degree of ZnO (NP) according to the reaction (5). In After hydrothermal, Zn(OH)2 gives larger amount of [Zn(OH)4]2−growth units which decomposed and finally nucleation was created to form the ZnO NR in the reaction (6). The ZnO nuclei and crystal nuclei were exhibiting fast growth orientation in (0001) crystal direction [34].
3 Results and discussion
3.1 Powder X-ray diffraction (PXRD)
The structural and crystalline phases of pure ZnO were identified by X-ray diffraction studies using Pro Penalty CAL with Cu-Kα radiation (1.5406 Å). Figure 2a, b shows the PXRD pattern of ZnO NPs and ZnO NRs. In the PXRD pattern, the hexagonal wurtzite structure of ZnO NPs and ZnO NRs phase (100), (002), (101), (102), (110), (103), (200), (112), (201) and (004) planes indexed to 31.61°, 34.27°, 36.15°, 47.41°, 56.48°, 62.82°, 66.30°, 67.92°, 69.06°, 72.46° and 31.71°, 34.35°, 36.25°, 47.51°, 56.56°, 62.92°, 66.30°, 67.92°, 69.06°, 72.46° well matched with JCPDS NO: 36-1451 [36]. From the Fig. 2a, b, it is observed that, there were small changes in the lattice constant due to the shift of diffraction peaks in ZnO NRs from its normal ZnO NPs. Also there were small shift towards the right side was observed in all six diffraction peaks for ZnO NRs can be attributed to the surface effect [37]. Here the large surface tension originated from the small size of the NPs can induce lattice distortion and hence decrease in lattice constant. The crystallite size was calculated by using Scherrer’s formula;
where, λ is wavelength of the X-ray beam, θ is diffraction angle, D is crystallite size, K is shape factor and β is Full Width Half Maximum (FWHM) of the peak. The average grain size of ZnO NRs and ZnO NPs were found to be 27 and 24 nm respectively. The dislocation density (δ) of the ZnO NRs and ZnO NPs were estimated due to the defects presence in the sample which represents as the length of dislocation lines per unit volume of the crystal and is calculated by using the given relation
where D is the crystallite size. The dislocation density (δ) of the ZnO NRs and ZnO NPs is 17.36 × 10−4 and 13.71 × 10−4 nm−2 [38]. The strain was calculated due to the crystal imperfection and distortion induced in powders by using the Williamson–Hall (W–H) methods formula
From the strain, stress can be calculated by substituting the value of ε in the relation σ = Cε. According to Hooke’s law, bulk Young’s modulus (C) = 1.46 ± 1010 N/m2 [38, 39]. The strain (ε) and stress (σ) of the ZnO NRs and ZnO NPs were found to be 1.12, 1.78% and 163, 259 MPa respectively. Figure 3 shows the wurtzite structure model of ZnO. From the Figure, it is seen that each type of atoms forms hexagonal-close-pack sub lattices and alternatively stacks along the c-axis. Each Zn2+ sub lattices contains four Zn2+ ions per unit cell and Zn2+ ions are surrounded by four O2− ions, or vice versa, which are coordinated at the edges of the tetrahedron. The non-central symmetric structure leads the piezoelectricity of ZnO, which makes it suitable for fabrication of various electronic devices and electromechanical sensors [40].

Powder XRD results of ZnO NPs and NRs

Schematic diagram of wurtzite structure of ZnO
3.2 UV–Visible spectroscopy
The presence of ZnO NPs and ZnO NRs was primarily confirmed by using UV–Visible (JASCO V650) spectroscopy technique. Figure 4a, b shows the proper changes in the optical properties between before and after Microwave-assisted hydrothermal of ZnO NPs and NRs. In before hydrothermal process the peak was observed around 368 nm and after hydrothermal process the peak was shifted to around 377 nm approximately observed from the figure. The observed shift in peak value is well coincide with the reported value [41] and the optical band gap of the before and after hydrothermal of ZnO NPs and NRs was calculated from the Tauc’s relation, and the optical absorption coefficient is given by Tauc’s relation as follows:
where hυ denotes the photon energy, Eg represents the band gap of the semiconductor and n is representing the nature of optical transition which may have direct, indirect and forbidden direct or forbidden indirect transitions which corresponds to the values of 1/2, 2, 3/2 or 3 respectively and α is the optical (or) absorbance coefficient defined by the Beer–Lambert law as follows:
where A denotes the absorbance and l denotes the optical path length respectively [42]. The direct band gap was estimated by the relation between (αhυ)1/2 and (hυ) as shown in Fig. 4b). Where h = (6.626 × 10−34 Js) represents the planks constant, λ denotes the wave length and (υ = c/λ) c = (3 × 108 m/s) is the velocity of light. The calculated band gap energy was found to be 3.52 and 3.32 eV respectively, depicting the presence of the direct band gap of the as-synthesized before and after Microwave-assisted hydrothermal of ZnO. Because of the effect of hydrothermal, 0.2 eV of the band gap energy was reduced and hence there is a small blue shift occurs in the material due to the quantum confinement effect. The occurrence of quantum confinement effect may be due to the chemical defects or vacancies present in the intergranular regions generating new energy level to reduce the band gap energy. The band gap decreases after hydrothermal of ZnO which indicates the possibility of the material being used in opto-electronic devices [19].

a UV–Visible spectra of ZnO before and after hydrothermal. b Direct energy band gap of ZnO before and after hydrothermal
3.3 Fourier transform infrared spectroscopy (FT-IR)
FT-IR analysis was taken for the samples in solid state using Perkin-Elmer spectrometer in the range of 400–4000 cm−1 to find the presence of various functional groups and the quality of the raw material. The FT-IR spectrum of ZnO NPs and ZnO NRs is shown in Fig. 5a, b. From the Figure, it is confirmed that a small stretch observed around 535 cm−1 indicating the bending vibration of Zn–O [43]. The broad band at 3400 cm−1 is attributed to –OH stretching vibrations, possibly including H2O, alcoholic OH, phenolic OH and/or carboxylic OH [44]. The peak around at 3188 cm−1 assigned to the N–H stretching vibration [31], indicates that a new complex was formed between ZnCl2 and N2H4. The peak around at 2923 and 2850 cm−1 is due to C–H symmetric and asymmetric stretching vibration [45] confirming that the molecules are not strongly adsorbed on ZnO crystal surface. The loss of band at 1624 cm−1 was due to the OH bending of water, which reveals the distinct dehydration of water molecule in the crystal lattice. Bending vibration of OH is appeared at 1100–1600 cm−1, which is attributed to the excessive hydroxylation of ZnO nanostructure [46]. The band at 1036 cm−1 is attributed to the C–N stretching vibration and the band at 717 cm−1 is due to lattice Zn–OH bending vibration in Zn(OH)2 [47, 48].

FTIR spectrum of ZnO NPs and NRs
3.4 FT-Raman spectroscopy
FT-Raman spectrum of ZnO NPs and NRs was recorded using a confocal Raman microscope (RENISHAW, United Kingdom) for the spectral range 300–600 cm−1 and shown in Fig. 6. The Raman signals are very sensitive to the structure of the crystalline material as well as to the defects in the crystal structure. The crystal structure of ZnO is wurtzite (hexagonal) with C6v-4(P63mc) point group symmetry and group theory predicts that there are two A1, two E1, two E2 and two B1 modes, where two B1 modes are Raman inactive and the other six modes are Raman active [49]. The dominant peak at 439 cm−1 is known as the Raman active dominant E2H mode of the wurtzite hexagonal phase of ZnO [50]. The peak at 334 cm−1 is the second order Raman spectrum originating from the zone boundary phonons 3E2H-E2L [49]. The small and broad peak with low intensity at 582 cm−1 attributed to the E1 (LO) mode. Generally, it is believed that the E1 (LO) mode is related to the structural defects (oxygen vacancies, zinc interstitials, free carriers, etc.) in ZnO [51].

Raman spectrum of ZnO NPs and NRs. (Color figure online)
3.5 Photoluminescence (PL) spectroscopy
The PL spectrum of ZnO NPs and NRs were carried out using Horiba Spectroflourometer. The PL spectrum of ZnO NPs and NRs is shown in Fig. 7a, b. From the Figure, it is observed that the samples exhibit two emission peak such as Near-Band-Edge emission (NBE) at 384 nm (∼3.2 eV) and broad Deep Level visible Emission (DLE) ranging from 425 to 625 nm with a band maximum at 512 nm, which is attributed to the recombination between the electrons in a deep defect level or a surface defect level and the holes in the valence band [49, 52]. The NBE emission has been reported to originate from the direct recombination of excitons and the visible emission from the radiative recombination from different defect states like oxygen vacancies (Vo), zinc vacancies (VZn), zinc interstitials (Zni), oxygen interstitials (Oi) or impurity levels present in the samples which depending on their preparation conditions [52]. Blue shift of UV peak was observed in the case of ZnO NRs compared with the prepared bulk ZnO NPs. During the hydrothermal process, if the temperature increases the DL emission intensity increases due to the lose of oxygen molecule [37, 53]. This reaction proceeds the NBE gradually increases with decrease in visible emission with the sample and hence exhibiting maximum NBE. This change may be due to the formation of crystalline ZnO rods with reduced surface defects by the gradual decomposition of hydroxide precursor with time [52]. The existence of doubly ionised oxygen vacancies as surface defects plays an important role in the enhanced photocatalytic activity of ZnO rods.

Photoluminescence (PL) spectra of ZnO before and after hydrothermal. (Color figure online)
3.6 Brunauer–Emmett–Teller (BET) analysis for ZnO NRs and ZnO NPs
The BET analysis is used to find out the presence of nitrogen adsorption and desorption of the synthesized material. The BET analysis of ZnO NPs and ZnO NRs are shown in the Fig. 8a, b. From the Figure, the maximum volume of adsorption and desorption were observed from ZnO NRs than ZnO NPs. At higher pressure the adsorption was increased owing to its capillary condensation in pores and also it happened by the surface modification of the sample and desorption happened by the modification in their pores texture [41, 54]. The BET surface area and total pore volume of ZnO NPs and ZnO NRs was found to be 20.84, 27.82 m2/g and 0.044, 0.071 cc/g respectively. The result shows that the values of surface area and total pore volume of ZnO NPs is very less compared to ZnO NRs which may be due to the surface modification of the material. Also confirms the mesoporous nature of the material by obtaining H3 type hysteresis loop [41].

BET analysis of a ZnO NPs and b ZnO NRs
3.7 Field emission scanning electron microscopy (FESEM)
The formation of ZnO NPs and NRs were confirmed (before and after Microwave-assisted hydrothermal) respectively from the FESEM images using Carl Zeiss microscopy Ltd, UK & SIGMA. Figure 9a, b shows the surface morphology of ZnO NPs and Fig. 9c, d shows the surface morphology of ZnO NRs respectively. FE-SEM analysis of the synthesized ZnO NPs reveals the agglomerated nature of spherical particle and ZnO NRs indicates the uniform distribution of rod like structure. The previous literatures have confirmed that the growth velocity of ZnO crystal in different orientations has discrepancies, which can be concluded as follow: > v (0001) > v (0110) > v (0001) [22]. Therefore, it can be concluded that the formation of the NRs may be due to the preferential growth along (0001) direction. The dimension of the ZnO NRs is shown in Fig. 9e, f. From the analysis, the minimum, median, maximum length and diameter of the ZnO NRs have been determined and the values are found to be 241.49, 646.73, 941.16 and 65.14, 122.18, 170.93 nm respectively. The standard deviation of ZnO NRs length and diameter was found to be 163.77 and 21.89 nm. The average length and diameter of the ZnO NRs size was very low compared to previous report by Tang et al. [55]. The EDAX analysis of ZnO (NPs) and ZnO (NRs) was shown in the Fig. 10a, b. From the EDAX spectrum, it is confirmed that, there are no impurities presents and only the elements Zn and O presents in the ZnO (NPs) and ZnO (NRs) samples.

Surface morphology of a, b ZnO NPs c, d ZnO NRs e, f particle size distribution of ZnO NRs length and width

EDAX spectrum of a ZnO NPs and b ZnO NRs. (Color figure online)
3.8 Thermal analysis
Analyzing the thermal stability of the material is an important parameter for the device fabrication [56]. Thermal stability of the ZnO NP’s and ZnO NR’s was studied by TGA and DTA analysis and the resultant thermogram is shown in Fig. 11a, b respectively using EXSTAR SII TG/DTA 6300 model. TGA analysis was made from 50 to 1000 °C with a heating rate of 10 °C/min under nitrogen atmosphere. From the results of TG analysis of ZnO NPs (before hydrothermal) weight loss of 1.02% was observed between 150 and 280 °C due to the decomposition of Zn(OH)2 [57]. Continuously a weight loss of 2.97% occurred in the temperature range 181–321 °C representing the loss of OH− and CO3 2− molecules from the material [58] which is confirmed by the observed endothermic peak at 231 °C. The weight loss of 4.37% was occurred at 405 °C, which may be due to the possible decarbonation process of the ZnO NPs at higher temperature [59]. The corresponding peak observed in DTA curve around 348 °C confirms the decomposition of CO2 and H2O gases from the ZnO [60]. From the Fig. 9b, (after hydrothermal) it is observed that a weight loss between 150 and 280 °C due to the decomposition of Zn(OH)2 and in the temperature range 280–352 °C corresponding to loss of (16.94%) and this could be attributed to decomposition of CO2 and H2O gases from the ZnO [57, 60] which is also confirmed by the sharp peak observed at 321 °C in DTA curve. Thus, it can be concluded from TG–DTA curves that, no thermal effect and no further weight loss were observed at temperature range 450–600 °C indicating that the decomposition does not occur above 450 °C and the stable remaining residues were may be atributed to ZnO [58].

TG and DTA thermogram of synthesized a ZnO NPs and b ZnO NRs
3.9 Zeta potential analysis
Figure 12a, b shows the zeta potential of the synthesized ZnO NPs and NRs. The analysis was taken on the samples in liquid state by using the instrument Zeta sizer nano-series (Malvern). Here the incident laser beam induces the Brownian movement of synthesized nanoparticles. Zeta potential shows high repulsive force and polydispersity between the particles [61]. The magnitude of the zeta potential between − 30 to + 30 mV shows the potential stability of the colloidal solution [62]. The colloidal solution of ZnO NPs and NRs can form agglomerates or aggregates. Agglomerate is a group of primary particles gathered by weak van der Waals forces and aggregate is referred to as a lump of primary particles held by the strong chemical bonds [63]. The hydrodynamic size of the ZnO NPs suggested that ZnO formed small aggregates when dispersed in aqueous condition [64]. The stability of the ZnO NRs and NPs were determined to be − 17.2 and − 31.4 mV respectively.

Zeta potential distribution of a ZnO NPs and b ZnO NRs
3.10 Dielectric properties
The frequency dependence of the dielectric response in material reveals information about the electric field distribution and charge transport mechanism which is correlated with electro-optic properties of the material and suitability of the material for electro optic applications. Significantly, new materials with low dielectric constant are the essential parameters for the microelectronics industry since, the material acts as an Inter Layer Dielectrics (ILD) [65]. The different polarization mechanisms and electrical processes that took place in the material has been confirmed by the study of the dielectric parameters such as dielectric loss and dielectric constant as a function of frequency and at different temperatures using HIOKI 3532 LCR HITESTER for the frequency range from 1 to 1 MHz. The ZnO NRs are made as pellet using hydraulic pressure. The pelletized sample was electroded on both sides with high purity silver paste which act as a parallel plate capacitor. The variation of dielectric constant of ZnO NRs with respect to frequency and temperature reveals that the dielectric constant decreases with increasing frequency and finally it becomes almost a constant at higher frequencies for all temperatures which is shown in Fig. 13a, b. It is also to be noted that dielectric constant increases with increase in temperature. Generally, dielectric behavior depends on the type of polarization mechanism happens inside the sample for a given frequency. The greater polarizability of the molecule leads to a higher dielectric constant of the material. When an alternating field is applied the dipoles in the material will be continually switching its position to reorient with the field. If the frequency of the field increases due to their inertia, the dipoles cannot maintain with the field and the alternation of the polarization will be lag behind the field. This corresponds to a reduction in the apparent polarization created by the field, which appears in measurements as an apparent reduction in the permittivity of the material therefore dielectric constant decreases [66]. The value of dielectric constant decreases up to 3 Hz in the nano material, which may be due to the presence of space charge polarization and the gradual increases of dielectric constant below 1 Hz are may be due to the electronic polarization. Space charge polarization is caused due to the presence of surface defects and vacancies in the material and these surface defects results in change of space charge distribution at the interfaces. On applying electric field the space charges moves and trapped in the defects [67]. On increasing frequency the space charge polarization decreases because the space charge does not follows the change in field. Therefore the dipoles are also incapable to follow the varying electric field [68]. This causes the decrease of dielectric constant value with increase in temperature at low frequencies. Figure 13c, d shows the variation of dielectric loss with frequency and temperature. The dielectric loss decreases with increase in frequency. However, Dielectric loss is the dissipation of energy through the movement of charges in a discontinuous electromagnetic field as polarization switches direction. This lagging polarization is caused by the presence of defects in the material [69, 70]. In figure the low dielectric loss value indicates that the material has minimum number of defects. Low value of dielectric constant and dielectric loss at higher frequencies makes the material as potential candidate for electro optic, microelectronic and telecommunication applications [71]. The dielectric constant and dielectric loss values of ZnO NRs were lower than the previously reported values [69].

Dielectric constant with respect to a frequency and b temperature for ZnO NRs and dielectric loss with respect to c frequency and d temperature for ZnO NRs
3.10.1 AC conductivity
AC conductivity measurement is important to study the electrical property of a material [72]. From the obtained dielectric values of ZnO NRs ac conductivity was calculated using the relation.
where εr is the relative permittivity, tanδ is loss tangent, εo is the vacuum dielectric constant (8.85 × 10−12 F/m). Figure 14a, b shows the ac conductivity of ZnO NRs for different frequency and temperature. It is observed from the Figure that the conductivity increases with increasing temperature and frequency. The minimum electrical conduction at lower temperature is governed by the impurities present in the material. At lower temperatures, energy needed to form the defect is greater than the energy needed for its drift [73, 74]. Whereas at high temperature more and more defects are created, the conductivity increases due to the creation of electronic pathways [75, 76]. Figure 14c shows the plot of σ ac versus 1000/T for the frequencies 1, 10, 100 kHz and 1 MHz. The variation of σ ac for different 1000/T values are found almost to be linear, the activation energy was calculated by fitting the curve using Arrhenius relationship
where σo is the pre exponent factor, E ac is the activation energy, K is the Boltzman constant and T is absolute temperature. The activation energy is obtained by using the relation
The slope value is calculated from the plot between σ ac and 1000/T. The activation energy obtained for the frequencies 1, 10, 100 kHz and 1 MHz are found to be 0.1686, 0.1671, 0.1655, 0.1216 eV respectively. The calculated activation energies are found decreasing with increase in frequency (Fig. 14d), which is due to the jumping of charge carriers between localized states [77]. Compare to the previous research works the ZnO NR material has fractional value of activation energy and low value of ac conductivity due to small amount of impurities presents in the synthesized material [78,79,80,81].

Variation of ac conductivity with respect to a frequency b temperature. c Plot of σ ac versus 1000/T for the frequencies of 1, 10, 100 KHz and 1 MHz for ZnO NRs. d Variation of activation energy with frequency
4 Conclusion
The synthesis of ZnO NPs and NRs without addition of any surfactant or complexing agent is reported in the present investigation. The Microwave-assisted hydrothermal technique demonstrated that the method is more convenient for the synthesis with desired nanostructures as compared to other techniques. The presence of various functional groups in the synthesized ZnO NPs and NRs were identified by FTIR analysis. Stretching of vibrational, rotational and low frequency transitions in molecules of ZnO nanostructures are confirmed by FT-Raman spectroscopy. The wurtzite hexagonal structure of ZnO nanostuctures is confirmed by the powder XRD analysis. The stability of the zeta potential distribution of ZnO NPs and NRs were found to be − 17.2 and − 31.4 mV respectively. The surface modification between ZnO NPs and ZnO NRs was clearly discussed by BET analysis. The thermal behaviour of the ZnO nanostructures was studied and found that no thermal effect and no further weight loss when increases the temperature above 450 °C. The maximum length and width of ZnO NRs were found to be 941.16 and 170.93 nm respectively for the temperature maintained around 160 °C for 45 min. Low value of dielectric constant and loss makes the material as efficient candidate for electro optic, electronic devices and telecommunication applications. Also the material fulfills the requirement of microelectronic device fabrication by having the small value of dielectric constant and fraction amount of dielectric loss. From all the above results, it is concluded that the ZnO rods are having an excellent property which is not only depending on the particle size, band gap and specific surface area but also on other factors such as morphology and surface defects.
References
K. Keem, H. Kim, G.T. Kim, J.S. Lee, B. Min, K. Cho, M.Y. Sung, S. Kim, J. Appl. Phys. Lett. 84, 4376 (2004)
M.S. Arnold, P. Avouris, Z.W. Pan, Z.W. Wang, J. Phys. Chem. B 107, 659 (2003)
M.H. Huang, S. Mao, H. Feick, H.Q. Yan, Y.Y. Wu, H. Kind, E. Weber, R. Russo, P.D. Yang, J. Sci. 292, 1897 (2001)
C. Lee, J,T.J. Lee, S.C. Lyu, Y. Zhang, H. Ruh, H.J. Lee, J. Appl. Phys. Lett. 81, 3648 (2002)
W.I. Park, Y.H. Jun, S.W. Jung, G.C. Yi, J. Appl. Phys. Lett. 82, 964 (2003)
G.C. Yi, C. Wang, W.I. Park, J. Semicond. Sci. Technol. 20, S22 (2005)
J.C. Tao, X. Chen, Y. Sun, Y. Shen, N. Dai, J. Colloids Surf. A 330, 67 (2008)
K.R. Reddy, V.G. Gomes, M. Hassan, Mater. Res. Exp. (2014). https://doi.org/10.1088/2053-1591/1/1/015012
M. Cakici, K. Raghava Reddy, F.A. Marroquin, Chem. Eng. J. (2016). https://doi.org/10.1016/j.cej.2016.10.012
Y.F. Zheng, X.J. Yu, X.Q. Xu, D.L. Jin, L.H. Yue, J. Ultrason. Sonochem. 17, 7 (2010)
J.P. Liu, X.T. Huang, Y.Y. Li, J.X. Duan, H.H. Ai, J. Mater. Chem. Phys. 98, 523 (2006)
Z.L. Wang, J. Mater. Today 26, 337 (2004)
J.H. Park, H.J. Choi, Y.J. Choi, S.H. Sohn, J.G. Park, J. Mater. Chem. 14, 35 (2004)
K.R. Reddy, K. Nakata, T. Ochiai, T. Murakami, D.A. Tryk, A. Fujishima, J. Nanosci. Nanotechnol. 11, 3692 (2011)
R.K. Joshi, J.J. Schneider, J. Chem. Soc. Rev. 41, 5285 (2012)
A.M. Showkat, Y.P. Zhang, M.S. Kim, A.I. Gopalan, K. Raghava Reddy, K.P. Lee, Bull. Korean Chem. Soc. 28, 1985 (2007)
A.C. Cakir, S. Erten-Ela, J. Adv. Powder Technol. 23, 655 (2012)
H.S. Hafez, Phys. E 44, 1522 (2012)
K. Ocakoglu, S.A. Mansour, S. Yildirimcan, A.A.A. Ghamdi, F. El-Tantawy, F. Yakuphanoglu, J. Spectrochim. Acta A 148, 362 (2015)
W.L. Suchanek, J. Cryst. Growth 312, 100 (2009)
B. Liu, H.C. Zeng, J. Am. Chem. Soc. 125, 4430 (2003)
H. Zhang, D. Yang, Y. Ji, J. Phys. Chem. B 108, 3955 (2004)
L.L. Kelly, D.A. Racke, H. Kim, P. Ndione, A.K. Sigdel, J.J. Berry, S. Graham, D. Nordlund, O.L.A. Monti, Adv. Mater. 28, 3960 (2016)
Y.P. Zhang, S.H. Lee, K. Raghava Reddy, A.I. Gopalan, K.P. Lee, J. Appl. Polym. Sci. 104, 2743 (2007)
H. Wei, Y. Wu, N. Lun, C. Hu, J. Mater. Sci. Eng. A 393, 80 (2005)
J. Liang, J. Liu, Q. Xie, J. Phys. Chem. B 109, 9463 (2005)
U. Pal, P. Santiago, J. Phys. Chem. B 109, 15317 (2005)
L. Wu, Y. Wu, W. LU, Phys. E 28, 76 (2005)
X.M. Sun, X. Chen, Z.X. Deng, J. Mater. Chem. Phys. 78, 99 (2003)
J. Du, Z. Liu, Y. Huang, J. Cryst. Growth 280, 126 (2005)
H. Zhu, D. Yang, H. Zhang, J. Inorg. Mat. 42, 1210 (2006)
L.Y. Meng, B. Wang, M.G. Ma, K.L. Lin, Mater. Today Chem. 1–2, 63 (2016)
S. Ghosh, J. Chakraborty, Mater. Res. Expr. 3, 125004 (2016)
Y.X. Wang, J. Xu, X.G. Cheng, H.F. Xu, L.J. Liu, Appl. Mech. Mater. 110, 547 (2011)
S. Cho, S.H. Jung, K.H. Lee, J. Phys. Chem. C 112, 12769 (2008)
K. Shingange, G.H. Mhlongo, D.E. Motaung, O.M. Ntwaeaborwa, J. Alloys Compd. 657, 917 (2016)
V. Prasad, C. D’Souza, D. Yadav, A.J. Shaikh, N. Vigneshwaran, J. Spectrochim. Acta A 65, 173 (2006)
P. Bindu, S. Thomas, J. Theor. Appl. Phys. 8, 123 (2014)
K. Vallal Peruman, M. Mahendran, S. Seenithurai, R. Chokkalingam, R.K. Singh, V. Chandrasekaran, J. Phys. Chem. Solids 71, 1540 (2010)
Z. Peng, G. Dai, W. Zhou, P. Chen, Q. Wan, Q. Zhang, B. Zou, J. Appl. Surf. Sci. 256, 6814 (2010)
D. Ramimoghadam, M.Z. Bin Hussein, Y.H. Taufiq-Yap, Chem. Cent. J. 7, 136 (2013)
H. Chauhan, M.K. Singh, S.A. Hashmi, S. Dek, RSC Adv. 5, 17228 (2015)
Y. Lai, M. Meng, Y. Yu, X. Wang, T. Ding, J. Appl. Catal. B: 105, 335 (2011)
P.L. Ascough, M.I. Bird, S.M. Francis, T. Lebl, J. Archaeol. Sci. 38, 69 (2011)
J. Xie, Y. Li, W. Zhao, L. Bian, Y. Wei, J. Powder Technol. 207, 140 (2011)
A. Ashar, M. Iqbal, I.A. Bhatti, M.Z. Ahmad, K. Qureshi, J. Nisar, I.H. Bukhari, J. Alloys Compd. 678, 126 (2016)
M.V. Vaishampayan, I.S. Mulla, S.S. Joshi, J. Mat. Res. Bull. 46, 771 (2011)
A. Sh. Sohrabnezhad, Seifi, J. Appl. Surf. Sci. 386, 33 (2016)
M. Khan, C. Wei, M. Chen, J. Tao, N. Huang, Z. Qi, L. Li, J. Alloys Compd. 612, 306 (2014)
Y. Dai, Y. Zhang, Y.Q. Bai, Z.L. Wang, J. Chem. Phys. Lett. 375, 96 (2003)
Z.L. Wang, J. Phys. 16, R829 (2004)
S. Mukhopadhyay, P.P. Das, S. Maity, P. Ghosh, P.S. Devi, J. Appl. Catal. B 165, 128 (2015)
P. Uthirakumar, C.H. Hong, Mater. Charact. 60, 1305 (2009)
M. Hassan, E. Haque, K. Raghava Reddy, A.I. Minett, J. Chen, V.G. Gomes, Nanoscale 6, 11988 (2014)
L. Tang, X.B. Bao, H. Zhou, A.H. Yuan, Phys. E 40, 924 (2008)
V. Kannan, R. Rakhikrishna, J. Philip, S. Brahadeeswaran, J. Therm. Anal. Calorim. 116, 339 (2013)
K.G. Chandrappa, T.V. Venkatesha, K. Vathsala, C. Shivakumara, J. Nanopart. Res. 12, 2667 (2010)
D. Raoufi, Luminescence 134, 213 (2013)
F. Hassan, M.S. Miran, H.A. Simol, M.A.B.H. Susan, M.Y.A. Mollah, J. Bangladesh J. Sci. Ind. Res. 50, 21 (2015)
M.Y. Nassar, M.M. Moustafa, M.M. Taha, RSC Adv. 6, 42180 (2016)
R. Suntako, J. Appl. Mech. Mater. 481, 60 (2014)
N. Senthilkumar, E. Nandhakumar, P. Priya, D. Soni, M. Vimalan, I.V. Potheher, New J. Chem. 41, 10347 (2017)
K.M. Kim, T.H. Kim, H.M. Kim, H.J. Kim, G.H. Gwak, S.M. Paek, J.M. Oh, J. Toxicol. Environ. Health. Sci 4, 121 (2012)
W.S. Cho, B.C. Kang, J.K. Lee, J. Jeong, J.H. Che, S.H. Seok, J. Part. Fibre Toxicol. (2013). https://doi.org/10.1186/1743-8977-10-9
P. Gunter, J. Opt. Commun. 11, 285 (1974)
I. Latif, E.E. AL-Abodi, D.H. Badri, J. Al Khafagi, Am. J. Polym. Sci. 6, 135 (2012)
M. Soosen Samuel, J. Koshy, A. Chandran, K.C. George, J. Phys. B 406, 3023 (2011)
N. Senthil Kumar, M. Ganapathy, S. Sharmila, M. Shankar, M. Vimalan, I. Vetha Potheher, J. Alloys Compd. 703, 624 (2017)
M.L. Dinesha, G.D. Prasanna, C.S. Naveen, H.S. Jayanna, Indian J. Phys. 87, 147 (2013)
G.H. Cross, A.R. Oldroyd, M.R. Worboys, J. Opt. Commun. 74, 425 (1990)
N.Y. Maharani, A. Cyrac Peter, S. Gopinath, S. Tamilselvan, M. Vimalan, I. Vetha Potheher, J. Mater. Sci. 27, 5006 (2016)
S. Perumal, C.K. Mahadevan, J. Phys. B 367, 172 (2005)
M. Vimalan, T. RajeshKumar, S. Tamilselvan, P. Sagayaraj, C.K. Mahadevan, J. Phys. B 405, 3907 (2010)
T.P. Srinivasan, R. Indhirajith, R. Gopalakrishnan, J. Cryst. Growth 312, 542 (2010)
J. Benet Charles, F.D. Ganam, J. Cryst. Res. Technol. 29, 707 (1994)
R.C. Miller, J. Appl Phys Lett 5, 17 (1964)
R. Ertuğrul, A. Tataroğlu, Chin. Phys. Lett. 7, 077 (2012)
J.F. Cordaro, Y. Shim, J.E. May, J. Appl. Phys. 60, 4186 (1986)
A. Rohatgi, S.K. Pang, T.K. Gupta, W.D. Straub, J. Appl. Phys. 63, 5375 (1988)
Y. Shim, J.F. Cordaro, J. Am. Ceram. Soc. 71, 184 (1988)
G. Garcia-Belmonte, J. Bisquert, F. Fabregat-Santiago, Solid State Electron. 43, 2123 (1999)
Acknowledgements
The first author sincerely thanks to TEQIP – II for the financial support to carry out the work. The corresponding author acknowledges the TEQIP– II for providing the seed money to carry out the research work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Senthilkumar, N., Vivek, E., Shankar, M. et al. Synthesis of ZnO nanorods by one step microwave-assisted hydrothermal route for electronic device applications. J Mater Sci: Mater Electron 29, 2927–2938 (2018). https://doi.org/10.1007/s10854-017-8223-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-8223-5