
Abstract
Zinc oxide (ZnO) nanorods (NRs) were obtained by oxidation of Zn thin layers, prepared by thermal evaporation method under electric field, at different times of 2.5, 5, 7.5 and 10 h. The crystal structure of samples was investigated using X-ray diffraction (XRD) measurement, which showed samples had a wurtzite hexagonal structure. In addition, field emission scanning electron microscopy (FESEM) indicated that with an increase in the oxidation time, the NRs diameter increased too. Elemental analysis of samples was performed using energy-dispersive X-ray spectroscopy (EDX) which clearly confirmed the presence of Zn and O with an atomic ratio of 1:1. Further, Raman spectra of samples verified wurtzite hexagonal phase due to high- and low-frequency vibration of E2 modes at room temperature and showed that with an increase in the oxidation time, the lattice defects decreased whereas crystal quality of samples increased. Optical properties of samples were determined by UV spectroscopy and demonstrated that when the oxidation time was added, the band gap energy of ZnO NRs decreased, which was attributed to the increase in crystallite size.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Zinc Oxide (ZnO) is an n-type semiconductor material with direct and wide band gap 3.37 eV and a large exciton binding energy of 60 meV at room temperature [1, 2]. It is also a low cost, abundant, chemically stable, and nontoxic material [3] that is crystallized in two forms of hexagonal wurtzite-type and Zinc-blende [4].
In recent years, ZnO nanostructures have been of great interest due to their optics, electronics, photonic, optoelectronics, physical and chemical properties depending on their size, form and morphology [5, 6]. These properties lead to using ZnO in various applications including UV laser [7], light-emitting diodes [8], solar and dye-solar cells [9], nanogenerators [10], gas sensors [11], photodetectors [12], photocatalysts [13], field effect transistor [14], and nanolasers [15].
Nowadays, several methods are used to synthesize ZnO nanostructures, such as vapor liquid solid (VLS) [16, 17], pulsed laser deposition (PLD) [8], sol–gel [18], hydrothermal [16], chemical vapor deposition (CVD), metal organic chemical vapor deposition (MOCVD), and physical vapor deposition (PVD) [19, 20]. The results of these methods can lead to producing nanoparticles, thin film, nanoroads, nanowires, and nanobelts. Physical vapor deposition is a simple and low-cost repeatable method to produce ZnO nanostructures.
In recent decades, most research groups have investigated properties of ZnO nanostructures that are prepared by physical vapor deposition. O. A. Fouad deposited ZnO thin film by thermal evaporation method and investigated the properties of photo-catalyst activity [21]. In his study, M. S. Islam fabricated Zinc Oxide photo-electrode using modified thermal evaporation method for water splitting application [22]. In addition, M. G. Farag’s team conducted studies on optical and structural properties of thermal evaporated Zinc Oxide thin films on polyethylene terephthalate (PET) substrates [23]. S. Palimer et al. prepared undoped ZnO thin films by thermal evaporation method and investigated their structural, optical and electrical properties [24]. As the literature evinces, no electric field has been used for preparation of ZnO nanostructures.
Jouya et al. studied the effect of electric field on the growth of thermally evaporated Zn thin films [25]. Considering the positive effect of applied electric field on the growth of the Zn thin films, the layers prepared with thermal evaporation method under applying a strong electric field were considered for producing ZnO NRs in this study.
Oxidation of Zn thin films was carried out at the temperature of 550 °C in different times. Then the effect of oxidation time on physical characteristics of NRs was studied.
2 Experimental procedures
To deposit Zn thin layer, Zn granules with 99% purity were used as the primary material. The Zn thin film was deposited on the glass substrate by thermal evaporation method under the pressure of about 1.33 × 10−3 Pa. To use Zn layers with higher crystallinity for preparing ZnO NRs, Zn layers were deposited by applying a strong electric field. The well-cleaned glass substrates were held under an aluminum plate; molybdenum crucible was also used for evaporating of granules. To apply the electric field, a metal net was placed between the source and substrate. The positive and negative poles of voltage were connected to the aluminum plate and the metal net, respectively. In this case, an electric field of 134,000 V/m (up to down) was applied and Zn thin films were deposited on the substrates.
To obtain ZnO NRs, the Zn thin films were placed on a small size heater (handmade) with a temperature of 550 °C. The oxidation process of Zn thin films was carried out in the air at the times of 2.5, 5, 7.5 and 10 h.
Furthermore, the effect of the oxidation time on properties of ZnO NRs was studied. Surface morphology and crystal structural of the samples were investigated by SEM images and X-Ray Diffraction (XRD). Elemental composition and chemical structure of samples were checked by Energy-Dispersive X-ray spectroscopy (EDX) and Raman Spectroscopy, respectively. In addition, UV–visible spectroscopy was used to investigate the optical properties of these samples. The ZnO NRs samples were named S1, S2, S3 and S4 for the oxidation times of 2.5, 5, 7.5 and 10 h, respectively.
3 Results and discussion
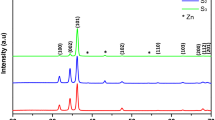
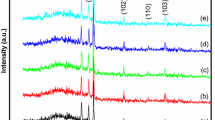
X-ray diffraction spectra of samples were recorded in a 2θ range of 20–71 degrees so as to analyze the crystal structure and to determine lattice parameters, the crystallite size, and lattice strain (Fig. 1). The peaks of (100), (002), (101), (102), (110), (103), (200), (112) and (201) show a wurtzite hexagonal crystal structure with P63-mc space group for all samples (JCPDS No: 01–089-0510). In addition to ZnO peaks in Samples S1, S2, and S3, Zn peaks can also be seen which shows Zn thin layer is incompletely oxidized. As the oxidation time increased, the intensity of these peaks decreased and eventually disappeared in Sample S4, which means the deposited Zn layer was completely oxidized.

XRD patterns of samples
The crystallite size of ZnO NRs was obtained using Debye–Scherrer formula [26]:
where D is the crystallite size, K is Scherrer constant, λ is the X-ray wavelength, β is the full width at half maximum (FWHM) of diffraction peak and θ is the Bragg angle.
Lattice parameters a and c were calculated by the following equation:
Where h, k and l are the Miller indexes, and d is the Bragg planes distance [27].
The Lattice strain (ε) and dislocation density (δ) were estimated using the Eqs. (3) and (4), respectively [28, 29].
The obtained results for single peaks of ZnO NRs, as the host structure, and for Zn as the guest structure, are listed in Tables 1 and 2, respectively. Also, the average values of crystallite size, strain, dislocation density, and lattice parameters of each sample were summarized in these Tables.
As it can be seen, the peak at (200) appears in Samples S3 and S4 in the ZnO structure, i.e. improving the oxidation process and increasing the crystalline order. Increasing the number of ZnO structure peaks indicates an increase in its crystallinity. Similarly, the increase in the peaks intensity in Fig. 1 also signifies this fact. However, the number of peaks related to Zn crystal structure dwindled with an increase in the oxidation time. The peak at 70.11 in Sample S3 disappeared, indicating that Zn crystallinity was lower than that of S1 and S2 Samples. The peaks related to the Zn structure have disappeared in S4, illustrating that all peaks belong to the ZnO structure.
The comparison of the peaks positions showed that in both ZnO and Zn structures no displacement occurred. The crystallite size for single peaks of a sample to another one did not have a significant variation. However, considering Fig. 1, the preferential growth direction of the sample has changed from (002) to (101). These changes are also observed in the FWHM values. As the oxidation time increased, the broadening of the peak (002) also increased from 0.402 to 0.431 whereas it increased from 0.432 to 0.453 for peak (101) and then decreased to 0.408. With these changes it can be concluded that the peak (101) in Sample S1 belonged to the Zn structure (corresponds to peak (002)), where by increasing the heating time and improving the oxidation process, the peak intensity reduced and then increased as the ZnO peak. However, the average crystallite size of ZnO NRs increased from 18.88 nm for Sample S1 to 23.11 nm for Sample S4, indicating a direct effect of heat on the ZnO NRs crystallinity. As the heating time increased, the sample oxidation improved, and the crystallinity in direction (200) was added to the sample. These changes in Zn structure occurred inversely. Thus, the average crystallite size of the Zn structure decreased from 31.25 nm for Sample S1 to 21 nm for Sample S3. Finally, in Sample S4, Zn structure disappeared, i.e. accomplishing the oxidation process of Zn thin layer. The comparison of lattice strain values for (002) and (101) peaks showed that as the oxidation time increased, the lattice strain increased and decreased, respectively, indicating improvement and diminution in the crystallinity in these directions. These variations confirm the change of preferential growth direction. In addition, the average of lattice strain showed that enhancement of the heating time led to a reduction in lattice strain. Furthermore, the dislocation density decreased with an increase in the crystallite size. Lattice strain and dislocation density of Zn structure increased, as expected, owing to the decrease in crystallinity and crystallite size. The comparison of lattice constants of ZnO structure showed that the a and c constants increased and decreased, respectively. These reveal that when the oxidation time increases, the ZnO hexagonal structure expands along the a-axis.
Figure 2 shows FESEM images of samples. In addition to the limited number of ZnO NRs in Sample S1, there are NRs in most of other locations which have started to grow. Therefore, in this Sample, remarkable areas of the Zn layer have not been oxidized yet. On the other hand, it confirms that the peak at 36.00 in XRD spectrum of Sample S1 is related to Zn layer. As the oxidation time increases, ZnO NRs grow in random directions and gradually become compressed (S2 and S3). Eventually, by improving the oxidation process, ZnO NRs grow with a relatively good order (S4).

FESEM images of the samples
Figure 3 illustrates histogram plots of ZnO NRs diameter distribution. Despite distributing the diameter sizes in a broad range (between about 20 and 80 nm) in Sample S1, the number of them is relatively limited indicating that the oxidation process is incomplete. In Sample S2, not only the size distribution of diameters has remarkably increased but also the number of NRs with diameter size between 40 to 50 nm has increased. This means that as the heating time increases, the rate of oxidation, and subsequently ZnO NRs crystallization, improves too. With an increase in the size distribution of the diameters as well as the number of NRs with a specific diameter, these changes continued in Sample S3. Finally, in Sample S4, the NRs diameter distribution became more limited and the number of NRs with a specific diameter (about 40–70 nm) increased significantly, indicating a more crystallinity and high-regular growth of ZnO NRs.

Histogram plots of NRs diameter size distribution
Table 3 shows ZnO NRs average diameter size for each sample obtained from Gaussian fit of histogram plots. To compare these results with those obtained from X-ray diffraction spectra, the XRD crystallite size and a constant have been listed in table too. As it can be seen, with an increase in the oxidation process time, the diameter of ZnO NRs increases too, indicating a progressive oxidation process and formation of thicker NRs. By comparing the FESEM and XRD results, it is concluded that an increase in the crystallite size and lattice parameter a confirms enhancing the diameter size obtained from FESEM analysis.
Elemental analysis of samples was performed using energy-dispersive X-ray spectroscopy. Figure 4 shows EDX spectra of the samples. The tables in the Figure also specify the atomic and weight percentages of each element in the compound. In all samples, EDX data are related to two elements of Zn and O, i.e. zinc layer has been oxidized. It also showed atomic ratio of Zinc in the range of 44–50% and oxygen in the range of 50–56%. These results are very close to the ratio expected for ZnO (1:1) [30, 31]. The comparison of Zn and O atomic percentages in the samples indicates that with an increase in the heating time, the oxidation process is more complete and a higher percentage of Zn layer is oxidized in agreement with XRD and FESEM analyses.

EDX analysis of the samples (insets show the weight and atomic percent of elements)
Raman spectra were recorded at room temperature to further investigate the structure of samples (Fig. 5). The optical phonons Γopt in the wurtzite crystals are given by Γopt = 1A1 + 2B1 + 1E1 + 2E2 in which the modes A1 and E1 are polar and are divided into transverse optical mode and longitudinal optical mode. E2 mode includes E2 (high) and E2 (low) frequency phonon modes. E2 (low) is related to the vibration of oxygen atoms and E2 (high) is related to heavy Zn sub-lattices. Also, B1 (low) and B1 (high) are not active Raman modes [32, 33]. According to Fig. 5, ZnO NRs peaks are seen at 331, 436, and 548 cm−1. The frequencies of Raman active phonon modes are listed in Table 4.

Raman spectra of the samples
Sharp characteristic peaks at 436 cm−1 with high-intensity related to E2 (high) mode and Raman second-order mode peak at 331 cm−1 indicate that the samples have a high crystalline wurtzite hexagonal phase [34]. The peak at 548 cm−1 corresponds to ZnO nanorods E1 (LO) mode due to lattice defects including oxygen vacancy and interstitial zinc [33,34,35]. Furthermore, the peak at 170 cm−1 is related to the glass substrate. As it can be seen, the peak intensity at 548 cm−1 decreases when the oxidation time increases and eventually disappears in Sample S4, which means lattice crystal defects decrease and the crystallinity increases, which is in agreement with lattice strain changes in the XRD analysis. In addition, when the heating time increases, the peak intensity at 436 and 331 cm−1 increase too, which indicates that crystal quality of samples has increased. Since peak at 436 cm−1 was the main characteristic peak in all samples, related FWHM values were measured and obtained 28, 24, 22, and 17 for the Samples S1, S2, S3 and S4, respectively. The results showed that from S1 to S4, the FWHM decreased which can be attributed to the increase of NRs diameter. Figure 6 shows the variation of FWHM of Raman peak at 436 cm−1 versus NRs diameter.

FWHM of Raman peak at 436 cm−1 vs. NRs diameter size
Figures 7 and 8 show optical properties including absorbance and transmittance spectra of samples in a wavelength range of 350–700 nm at room temperature. As it can be seen in absorption spectra of samples in Fig. 7, an absorption edge is observed around the wavelength of 372 nm. However, as the oxidation time increased, the optical absorption edge shifted to higher wavelengths (red shifts) due to an increase in crystallite size of samples. In addition, by increasing the heating time from S1 to S4, the optical absorption decreased, which is attributed to a reduction in the dislocations density and an increase in crystallite size. However, after 525 nm the optical absorption increased. These changes were reversed in transmittance spectra (Fig. 8).

Optical absorbance of the samples

Optical transmittance spectra of the samples
TAUC plots were used to calculate band gap energy of samples. This way, using optical absorption spectrum of samples, the αhν2 was plotted versus hν, then the tangent line was plotted on the linear region. The band gap energy was obtained by dividing the tangent line on the graph in the linear region and the hν axis at ahν2 = 0 (Fig. 9) [36]. The results showed that band gap energy in all samples were less than that for the bulk ZnO (3.37 eV [37]). In addition, as the oxidation time increased, band gap energy reduced, which is attributed to the increase in diameter size of NRs and the quantum confinement effects.

Plot of (αhν)2vs. hν to calculate band gap for samples S1–S4
4 Conclusion
A thin layer of Zn was deposited by Thermal Evaporation under an electric field. Then, Zn thin layer was exposed to the air to prepare ZnO NRs. To investigate the effect of oxidation time on ZnO NRs properties, Zn thin films were exposed to the air for 2.5, 5, 7.5 and 10 h. ZnO NRs crystal structure was investigated using X-ray diffraction spectra which showed samples had a wurtzite hexagonal crystal structure and as the oxidation time increased, the crystallite size increased too. The surface morphology of samples was recorded by scanning electron microscopy which showed that with an increase in heating time, the NRs diameter increased too. Energy-dispersive X-ray spectroscopy of samples clearly confirmed the presence of Zn and O with an atomic ratio of 1:1. Room temperature Raman spectra confirmed the wurtzite hexagonal phase of samples and showed that the crystallite size increased when the oxidation time was added. Investigation of optical properties showed that as the oxidation time increased, the absorbance and transmittance decreased and increased, respectively. The band gap energy of NRs also decreased with the heating time increase, which is attributed to the increase of diameter size and quantum confinement effects.
References
I. Wallace et al., Synthesis and characterization of zinc oxide (ZnO) nanowire. J Nanomed Nanotechnol 6(321), 2 (2015)
Y. Tong et al., Growth and optical properties of faceted hexagonal ZnO nanotubes. J. Phys. Chem. B 110(30), 14714–14718 (2006)
C. Dwivedi, V. Dutta, Size controlled synthesis of ZnO nanoparticles via electric field assisted continuous spray pyrolysis (EACoSP) reactor. Appl. Phys. A 109(1), 75–79 (2012)
F. Xue et al., Influence of external electric field on piezotronic effect in ZnO nanowires. Nano Res 8(7), 2390–2399 (2015)
Y. Zhang et al., Synthesis, characterization, and applications of ZnO nanowires. J. Nanomater. 2012, 20 (2012)
C.R. Chandraiahgari et al., Synthesis and characterization of ZnO nanorods with a narrow size distribution. RSC Adv. 5(62), 49861–49870 (2015)
S. Chu et al., Electrically pumped waveguide lasing from ZnO nanowires. Nat. Nanotechnol. 6(8), 506 (2011)
J.H. Na et al., Hybrid p-n junction light-emitting diodes based on sputtered ZnO and organic semiconductors. Appl. Phys. Lett. 95(25), 329 (2009)
P. Sudhagar et al., Facile synthesis of highly branched jacks-like ZnO nanorods and their applications in dye-sensitized solar cells. Mater. Res. Bull. 46(9), 1473–1479 (2011)
Z.L. Wang et al., Lateral nanowire/nanobelt based nanogenerators, piezotronics and piezo-phototronics. Mater. Sci. Eng. 70(3–6), 320–329 (2010)
J. Xu et al., Studies on alcohol sensing mechanism of ZnO based gas sensors. Sens. Actuat. B 132(1), 334–339 (2008)
C.-Y. Lu et al., Ultraviolet photodetectors with ZnO nanowires prepared on ZnO: Ga/glass templates. Appl. Phys. Lett. 89(15), 153101 (2006)
S. Cho et al., Large-scale fabrication of sub-20-nm-diameter ZnO nanorod arrays at room temperature and their photocatalytic activity. J. Phys. Chem. C 113(24), 10452–10458 (2009)
P. Li et al., Investigation on the mechanism of nanodamage and nanofailure for single ZnO nanowires under an electric field. ACS Appl. Mater. Interfaces 6(4), 2344–2349 (2014)
N. Morales-Flores et al., Morphology control and optical properties of ZnO nanostructures grown by ultrasonic synthesis. Adv. Nano Res. 1(1), 59–70 (2013)
Q. Zhou et al., Synthesis of vertically-aligned zinc oxide nanowires and their application as a photocatalyst. Nanomaterials 7(1), 9 (2017)
E.W. Petersen et al., Growth of ZnO nanowires catalyzed by size-dependent melting of Au nanoparticles. Nanotechnology 20(40), 405603 (2009)
G. Wu et al., Controlled synthesis of ZnO nanowires or nanotubes via sol–gel template process. Solid State Commun. 134(7), 485–489 (2005)
G. Jimenez-Cadena et al., Synthesis of different ZnO nanostructures by modified PVD process and potential use for dye-sensitized solar cells. Mater. Chem. Phys. 124(1), 694–698 (2010)
R. García-Gutiérrez et al., Luminescence and structure of ZnO grown by physical vapor deposition. Adv. Mater. Sci. Eng. 2012, 872597 (2012)
O. Fouad et al., Zinc oxide thin films prepared by thermal evaporation deposition and its photocatalytic activity. Appl. Catal. B 62(1–2), 144–149 (2006)
Islam, M., et al. Zinc oxide thin film fabricated by thermal evaporation method for water splitting application. In: 2015 International Conference on Electrical & Electronic Engineering (ICEEE). 2015. IEEE.
M. Faraj, K. Ibrahim, Optical and structural properties of thermally evaporated zinc oxide thin films on polyethylene terephthalate substrates. Int. J. Polym. Sci. 2011, 302843 (2011)
Palimar, S., G. Shivakumar, K.V. Bangera. Conductive and transparent undoped ZnO thin films prepared by thermal evaporation method. In: AIP Conference Proceedings. 2012. AIP.
M. Jouya, F. Taromian, S. Siami, Rapid growth of zinc oxide nanobars in presence of electric field by physical vapor deposition. J. Theor. Appl. Phys. 11(4), 291–299 (2017)
M. Afshari, M. Ghazi, M. Izadifard, Structural and magnetic properties of Fe/Cu/Fe trilayers. Afr. Rev. Phys. 10, 27–37 (2015)
M.H. Amerioun et al., Fabrication of CuInS2/CNTs absorber layers by sol–gel method. Mater. Sci. Semicond. Process. 38, 149–156 (2015)
M. DasariAyodhya et al., Synthesis, characterization of ZnS nanoparticles by coprecipitation method using various capping agents-Photocatalytic activity and Kinetic study. IOSR J. Appl. Chem. 6(1), 01–09 (2013)
S. Terohid et al., Effect of growth time on structural, morphological and electrical properties of tungsten oxide nanowire. Appl. Phys. A 124(8), 567 (2018)
S.N. Alam et al., SEM, EDX & XRD of zinc oxide nanostructures synthesized by zinc oxidation. Microsc. Anal. 26(4), 11–14 (2012)
A.F. Abdulrahman et al., Different substrates effects on the topography and the structure of the ZnO nanorods grown by chemical bath deposition method. Dig. J. Nanomater. Biostruct. 11(3), 1007–1016 (2016)
R. Zhang et al., Photoluminescence and Raman scattering of ZnO nanorods. Solid State Sci. 11(4), 865–869 (2009)
I. Musa, N. Qamhieh, S.T. Mahmoud, Synthesis and length dependent photoluminescence property of zinc oxide nanorods. Results Phys. 7, 3552–3556 (2017)
N. Neykova et al., Study of ZnO nanorods grown under UV irradiation. Appl. Surf. Sci. 472, 105–111 (2019)
Y. Zhang et al., Synthesis, characterization, and applications of ZnO nanowires. J. Nanomater. 2012, 624520 (2012)
Islam, M., et al. Zinc oxide thin film fabricated by thermal evaporation method for water splitting application. In 2015 International Conference on Electrical & Electronic Engineering (ICEEE). 2015. IEEE.
X. Li et al., Fabriction of ZnO nanorods with strong UV absorption and different hydrophobicity on foamed nickel under different hydrothermal conditions. Micromachines 10(3), 164 (2019)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jouya, M., Taromian, F. & Afshari Abolkarlou, M. Growth of Zn thin films based on electric field by thermal evaporation method and effect of oxidation time on physical properties of ZnO nanorods. J Mater Sci: Mater Electron 31, 8680–8689 (2020). https://doi.org/10.1007/s10854-020-03403-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-020-03403-w