
Abstract
Partition coefficients describe the equilibrium partitioning of a single, defined charge state of a solute between two liquid phases in contact, typically a neutral solute. Octanol–water partition coefficients (\(K_{\rm ow}\)), or their logarithms (log P), are frequently used as a measure of lipophilicity in drug discovery. The partition coefficient is a physicochemical property that captures the thermodynamics of relative solvation between aqueous and nonpolar phases, and therefore provides an excellent test for physics-based computational models that predict properties of pharmaceutical relevance such as protein-ligand binding affinities or hydration/solvation free energies. The SAMPL6 Part II octanol–water partition coefficient prediction challenge used a subset of kinase inhibitor fragment-like compounds from the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) prediction challenge in a blind experimental benchmark. Following experimental data collection, the partition coefficient dataset was kept blinded until all predictions were collected from participating computational chemistry groups. A total of 91 submissions were received from 27 participating research groups. This paper presents the octanol–water log P dataset for this SAMPL6 Part II partition coefficient challenge, which consisted of 11 compounds (six 4-aminoquinazolines, two benzimidazole, one pyrazolo[3,4-d]pyrimidine, one pyridine, one 2-oxoquinoline substructure containing compounds) with log P values in the range of 1.95–4.09. We describe the potentiometric log P measurement protocol used to collect this dataset using a Sirius T3, discuss the limitations of this experimental approach, and share suggestions for future log P data collection efforts for the evaluation of computational methods.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL) challenges (http://samplchallenges.github.io) are a series of blind prediction challenges for the computational chemistry community that aim to evaluate and advance computational tools for rational drug design [1]. These challenges focus the community on specific phenomena relevant to drug discovery—such as the contribution of force field inaccuracy to binding affinity prediction failures—and, using carefully-selected test systems, isolate these phenomena from other confounding factors. Through recurring community exercises involving blind prediction followed by data sharing and discussion, these challenges evaluate tools and methodologies prospectively, enforce data sharing to learn from failures, and generate high-quality datasets into the community as benchmark sets. As a result, SAMPL has driven progress in a number of areas over six previous rounds of challenge cycles [2,3,4,5,6,7,8,9,10,11,12,13,14,15].
To assess the accuracy of different computational methods, SAMPL has relied on the measurement of simple host-guest association affinities [6, 8, 11, 15,16,17,18,19] and other physical properties that isolate issues such as failing to capture relevant chemical effects, computationally-intensive conformational sampling, and force field accuracy. In SAMPL5, for example, a log D challenge was devised with the goal of isolating the accuracy of protein-ligand force fields from the difficulties of configurational sampling [20, 21]. In addition to being a useful surrogate for the accuracy of force fields in predicting binding free energies, partition or distribution coefficients are frequently used as a measure of lipophilicity in pharmacology [22], or as surrogates for solubility, permeability [23], and contributors to affinity [22, 24]. Lipophilicity is a critical physicochemical property that affects ADMET (absorption, distribution, metabolism, excretion, and toxicity) [22, 25, 26]. Since log P is utilized as a predictor for good drug-like properties in terms of pharmacokinetics and toxicity [25], accurate log P predictions of virtual molecules have high potential to benefit drug discovery and design.
Surprisingly, the cyclohexane-water log D challenge proved to be particularly problematic due to the necessity to account for protonation state effects to correctly compute the distribution coefficients, which assess the partitioning of all ionization states between phases [20]; failing to account for these protonation state effects led to modeling errors up to several log units [27]. As a result, the SAMPL6 Part II log P prediction challenge [28] aimed to further isolate the assessment of force field accuracy from the issues of conformational sampling and the modeling of ionization state equilibria by inviting participants to predict the partitioning of neutral drug-like molecules between aqueous and nonaqueous phasesFootnote 1. For maximum synergy with previous competitions, the challenge compound set was constructed to be a subset of kinase inhibitor fragment-like small molecules drawn from the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge set [29], where the accuracy of participants to predict \(\hbox {p}{K}_{{\rm a}}\) values was assessed. A blind challenge (the SAMPL6 Part II log P blind prediction challenge) was run from November 1, 2018 to March 22, 2019 in which participants were given molecular structures and experimental details and asked to predict octanol–water partition coefficients before the data was unblinded on March 25, 2019. All primary and processed data was made available at https://github.com/MobleyLab/SAMPL6 immediately following the close of the competition.
Partition coefficients and principles of their measurement
The partition coefficient describes the equilibrium partitioning of a molecule in a single, defined, charge state between two liquid phases in contact. Unless stated otherwise, in common usage partition coefficient (P or P0) refers to the partitioning of the neutral state of a molecule. In particular, the octanol–water partition coefficient of neutral species (frequently written as \(K_{ow}\) or P) is defined as
This quantity is often written in its \(\log _{10}\) form, which we denote here as log P,
However, ionic species can also partition between phases [30,31,32]. The partition coefficients of ionic species is calculated using the same equation, e.g.P+1 refers to the partition equilibrium of +1 charge state of a molecule. Based on the experimental measurement method this value may be defined for a single tautomer or may involve multiple tautomers.
A closely related concept is that of the distribution coefficient (\(D_{ow}\), often written in \(\log _{10}\) form as log D) which should not be confused with log P. log D is the logarithm of the sum of all species (both neutral and ionized) concentrations in the organic phase divided by the sum of neutral and ionic species concentrations in aqueous phase. Both octanol–water log P and log D values are frequently used as lipophilicity estimates [22]. However, while log D is pH-dependent, log P is independent of the pH of the aqueous phase. As log P is defined as the partition coefficient of neutral species, it would include all neutral tautomer populations if a compound can tautomerize.
The gold standard of partition coefficient measurement experimentation is the shake-flask method, according to the Organization for Economic Cooperation and Development (OECD) [33]. Methods developed as experimental refinements on the shake-flask method are high-throughput microscale shake flask [34, 35] and slow stirring methods [36]. Other direct methods for log P or log D determination include dialysis chamber-based methods [37], micellar electrokinetic capillary chromatography [38, 39], and counter-current chromatography [39]. An indirect experimental method that is widely used—despite being less reliable—is log P estimation based on reversed-phase high-performance liquid chromatography (HPLC) retention times [40,41,42,43,44]. The measurement principle for all of these methods is the measurement of log D—the equilibrium distribution coefficient for both neutral and ionized species—in a pH-dependent manner. As a result, in order to measure log P with these methods it is necessary to conduct the log D measurements at a pH where the analyte is completely un-ionized. At a pH where the analyte is at a neutral state, log P is equal to log D; however, accurately predicting or measuring the equilibrium ionization constant (\(\hbox {p}{K}_{{\rm a}}\)) of a substance is a prerequisite. Here in this study, however, we pursued an alternate approach for experimental determination of log P, which is potentiometric measurements.
Potentiometric measurement of log P with the Sirius T3
The potentiometric log P measurement method determines log P values directly using potentiometric titrations in an immiscible biphasic system [45, 46]. The shift of apparent \(\hbox {p}{K}_{{\rm a}}\) values when the aqueous phase is in contact with the octanol phase is used to estimate log P values. Experimental log P values presented in this study were collected using this potentiometric method, and they refer to the partition coefficient of the neutral species.

Potentiometric log P measurements are based on a model of ionization and partitioning equilibria [50]. Measurements of the \(\hbox {p}{K}_{{\rm a}}\) and apparent \(\hbox {p}{K}_{{\rm a}}\) (\(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)) at three octanol–water volumetric ratios (log R) are performed to estimate the partition coefficients of neutral and ionized species, log P0 and log P−1, respectively. An ionization and partitioning equilibria model, along with estimated potentiometric titration curves, are shown for a monoprotic acid in this figure
The potentiometric log P measurement method used by the Sirius T3 instrument (Pion) [46,47,48,49,50,51] is based on determination of the partition profile directly from acid-base titrations in a dual-phase water-partition solvent system consisting of two liquid phases in contact (Fig. 1). In this method, multiple potentiometric acid-base titrations are performed in the aqueous phase at various equilibrium volumetric ratios of octanol and water to observe the ionization and partitioning equilibrium behavior of the analyte. As the relative volume ratio of octanol to water changes, a shift in apparent \(\hbox {p}{K}_{{\rm a}}\) (\(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)) is observed due to partitioning of neutral and ionic species—which have distinct octanol–water partitioning equilibria—into the octanol-rich phase. Equations describing this coupled partitioning and ionization equilibria are then solved to determine the log P of the neutral and ionic species. To use this method, aqueous \(\hbox {p}{K}_{{\rm a}}\) value(s) must be known, and analytes must be fully water soluble at the highest concentration they reach during the titrations throughout the entire range of pH titration selected for the potentiometric log P measurement protocol. The largest pH range selected for titration can be pH 2–12 and the minimum range should include ±2 pH units around the \(\hbox {p}{K}_{{\rm a}}\) and \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\).
When an ionizable substance is titrated in a two-phase system, the apparent \(\hbox {p}{K}_{{\rm a}}\)—here, denoted \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)—observed in the titration shifts due to differential partitioning of neutral and ionized species into the nonaqueous phase. The \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) value is the apparent \(\hbox {p}{K}_{{\rm a}}\) in the presence of partition solvent octanol. Its shift is dependent on the volumetric ratio of the water and octanol phases. The \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) value increases with increasing partition solvent volume for monoprotic acids and decreases with monoprotic bases. The shift in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) is directly proportional to the log P of the compound and the ratio of octanol to water. For a monoprotic acid or base, the partition coefficient of neutral (P0) and ionic species (P−1, P+1) relates to \(\hbox {p}{K}_{{\rm a}}\) and \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) as [50],
Here, R is the volume ratio of nonaqueous phase (\(V_{\rm nonaq}\)) to aqueous phase (\(V_{\rm aq})\),
Methods
Compound selection and procurement
For the SAMPL6 Part II log P challenge, we attempted to collect log P measurements for the entire set of 24 kinase inhibitor fragment-like compounds selected for the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge [29, 52]. Details of compound selection criteria for the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) set—driven in large part by cheminformatics filtering for experimental tractability and rapid, inexpensive compound procurement—can be found in the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) experimental data collection paper [29]. Compounds with publicly available experimental log P measurements were excluded by checking the following sources: DrugBank [53], ChemSpider [54], NCI Open Database August 2006 release [55], Enhanced NCI Database Browser [56], and PubChem [57]. However, not all molecules selected for SAMPL6 were suitable for log P measurements using the Sirius T3, due to various reasons such as low solubility, apparent \(\hbox {p}{K}_{{\rm a}}\) value shifting out of experimental range, or log P values out of experimental range limited by the sample vial. These limitations are explained in more detail in the “Discussion” section. Only 11 small molecules proved to be suitable for potentiometric log P measurements.
Molecule IDs assigned to these compounds for the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) challenge were preserved in the SAMPL6 Part II log P Challenge. A list of SAMPL6 log P Challenge small molecules, SMILES, and molecule IDs can be found in Table 1. Counterions, where present in solid formulations (see “Potentiometric log P measurements” section below), were included in SMILES for the sake of completeness, although no significant effect is expected from the presence of chloride counterions as experiments were conducted using KCl to maintain constant ionic strength. Procurement details for all compounds in the SAMPL6 log P Challenge compounds are presented in Table S1.
Potentiometric log P measurements
Experimental octanol–water log P values of neutral species were collected using potentiometric log P (pH-metric log P) measurements [50] at 25.0±0.5 \(^\circ\)C and constant ionic strength (0.15 M KCl). Aqueous \(\hbox {p}{K}_{{\rm a}}\) values are required for log P determination with the Sirius T3, and were previously determined for all compounds in this set [29] using UV-metric \(\hbox {p}{K}_{{\rm a}}\) measurements [58, 59] with the same instrument.
Three independent replicates were performed for each log P measurement using 1-octanol and water biphasic systems at 25.0 \(^\circ\)C, starting with solid material. General guidance of according to the instrument manual suggests optimal analyte mass should be in the range of 1–10 mg. “Sample weight” is the terminology used to describe analyte mass in Sirius T3 manuals, software, and reports. Due to solubility limitations of the SAMPL6 compounds, we tried to use analyte masses less than 3 mg. There was not much flexibility to adjust aqueous phase volume, since this is limited by the minimum volume required for the pH probe (1.4–1.5 mL) and the volume that must be spared for the octanol phase in the sample vial. Therefore, we adjusted analyte mass instead of aqueous phase volume when reducing sample concentration was necessary to achieve solubility.
For molecules with low solubility, target analyte mass was reduced, but not below a minimum of 1 mg. Samples were prepared by weighing 1–3 mg of analyte in solid powder form into Sirius T3 analysis vials using a Sartorius Analytical Balance (Model: ME235P) equipped with an antistatic ionizer. It was difficult to transfer powder compounds to achieve target masses in 1–3 mg range exactly. Instead, we opted to weigh out approximate target mass (± 40% of the target mass was considered acceptable) and record the resulting sample mass. For instance, when aiming for 1 mg of compound, if 1.29 mg of compound was transferred to the balance, that was recorded as analyte mass and 1.29 mg was provided in to the Sirius T3 software for analysis. Reporting accurate analyte mass was important since analyte mass and purity are part of the Sirius T3 refinement model, although the analysis software doesn’t accept analyte purity as an input. Analyte purity (“sample concentration factor” according to Sirius T3) is estimated from the refinement model fit to experimental data given the reported analyte mass by the user. The remaining steps in sample preparation were performed by the automated titrator: addition of ionic-strength adjusted (ISA) water (typically 1.5 mL) and partition solvent (ISA water-saturated octanol), mixing, sonication, and titration with acid (0.5 M KCl) and base (0.5 M KOH) solutions targeting steps of 0.2 pH units. ISA water is 0.15 M KCl solution which was used to keep ionic strength constant during the experiment. ISA water was prepared by dissolving KCl salt in distilled water.
ISA water-saturated octanol was prepared by mixing 500 mL 1-octanol (Fisher Chemical, cat no A402-500, lot no 168525) with 26.3 mL ISA water (targeting 5% ISA water-octanol mixture by volume) and letting the mixture phases separate before attaching it to the automated titrator. Titrations were performed under argon flow on the liquid surface to minimize carbon dioxide absorption from the air.
In some cases, to help with kinetic solubility issues of the analytes, solid samples were predosed manually with 80–100 \(\upmu \hbox {L}\) ISA water-saturated octanol prior to the addition of ISA water and partition solvent—these are noted in Table 1. Predosed volumes were provided to the analysis software as an input and were accounted for in the total octanol volume calculation. Whenever mean molecular charge vs pH plots showed experimental data points that deviated from the expected sigmoidal curve shape (oscillatory shape or steeper descent), we suspected solubility problems and attempted to prevent them by predosing octanol, which can only help the cases in which the solubility issue is a kinetic and not an equilibrium solubility issue. The only way to alleviate an equilibrium solubility issue entirely is to lower the analyte concentration by starting the experiment with a smaller analyte mass.
For each replicate log P measurement, three sequential automated acid-base titrations were performed in the same vial at three different volume ratios of octanol and water, using 0.5 M KOH and HCl solutions as titrants while monitoring pH with a pH electrode (Ag/AgCl double-junction reference electrode). Additional volumes of octanol were dispensed before each titration to achieve target octanol–water ratios. The sequence of three octanol–water ratios were determined using predicted log R profiles (apparent \(\hbox {p}{K}_{{\rm a}}\) shift vs log\(_{10}\) of the volumetric ratios of partition solvents, as shown in Fig. 2c, d) or experimental log R profile if a previous iteration of the experiment is available during protocol optimization, with the goal of selecting three volumes that will maximize the |\(\hbox {p}{K}_{{\rm a}}\)- \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)| values between each titration. Experiments were designed so that maximum separation of \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values can be achieved while the total liquid volume in the analysis vial did not exceed 3 mL by the end of the third titration.
Two Sirius T3 software programs were used to execute measurement protocols (Sirius T3 Control v1.1.3.0) and analyze experiments (Sirius T3 Refine v1.1.3.0). The Sirius T3 Refine software has the capability of fitting partitioning and ionization equilibrium models to potentiometric data collected from a biphasic system to estimate log P values. The starting point for the model fit is simulated titration curves constructed using aqueous \(\hbox {p}{K}_{{\rm a}}\) values (using prior \(\hbox {p}{K}_{{\rm a}}\) measurements, here taken from [29]), predicted log P values, input analyte mass, and volumes of aqueous and organic phases dispensed to prepare the sample. Collected experimental measurements (pH vs dispensed volume of acid and base solutions) were used to refine the model parameters (log P of neutral species, log P of ionic species, analyte concentration factor, carbonate content, acidity error) to determine the log P values of neutral species and ions [48]. Potentiometric log P measurements have the potential to determine the partition coefficients of the ionic species (log P1) in addition to log P of the neutral species (log P0). It was, however, very challenging to design experiments to capture log P values of the ionic species due to volumetric limitations of the glass analysis vial and measurable pH range. Therefore, while optimizing experimental protocols, we prioritized the accuracy for only log P of the neutral species. Experimental protocols were optimized iteratively by adjusting octanol–water ratios, analyte concentration, and pH interval of the titration.
A partitioning and ionization equilibrium model [48] was fit to potentiometric measurements to estimate log P values of the neutral species and also the charged species, as implemented in Sirius T3 Refine Software. Experiments were optimized to determine log P of neutral species with good precision. log P estimates of charged species had high variance between replicate experiments performed in this study and were judged to be unreliable. Optimizing experiments further to be able to capture log P values of ionic species accurately would require larger log R values, which was limited by sample vial volume. Therefore, we decided not to pursue experimental data collection for ionic partition coefficients further.
Reporting uncertainty of log P measurements
Experimental uncertainties of log P measurements were reported as the standard error of the mean (SEM) of three or four replicates. The standard error of the mean (SEM) was estimated as
where \(\sigma\) denotes the unbiased sample estimator for the true standard deviation and \(\mu\) denotes the sample mean. \(x_i\) are observations and N is the number of observations.
The SEM calculated from independent replicate experiments as above was found to be larger than non-linear fit error reported by the Sirius T3 Refine Software from potentiometric log P model fit of a single experiment, thus leading us to believe that running replicate measurements and reporting mean and SEM of log P measurements better captured all sources of experimental uncertainty. We caution, however, that the statistical error estimated from three replicates is only determined to an order of magnitude [60].
Quality control of analytes
Purities of all SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge compounds—a subset of which formed the log P set used here—were determined by LC-MS and reported elsewhere [29]. The same lots of compounds were used for \(\hbox {p}{K}_{{\rm a}}\) and log P measurements. LC-MS assessment showed that the 11 compounds reported in this study have a minimum of 96.5% purity and matching molecular weight to supplier reported values (Table S1).
When questions were raised about the accuracy of log P measurements for SM13 by a participant of SAMPL6 log P Challenge, we had additional quality control experiments performed to confirm the compound identity of SM13. LC-MS and NMR data were fully consistent with the structure of SM13 as originally provided (Figure S1, S2). High-Resolution Mass Spectrometry (HRMS) data was acquired using an Agilent 6560 Q-ToF by +ESI. NMR data were acquired for the sample dissolved in pyridine-d5. 1H, DQF-COSY, and ROESY spectra were acquired using a 600 MHz Bruker AVANCE III HD spectrometer equipped with a liquid nitrogen-cooled broadband Prodigy probe. Chemical shifts were assigned to validate the structure of SM13.
Results
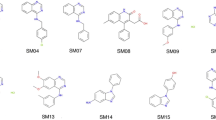
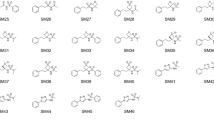
In this study, we attempted to use the potentiometric log P measurement method of the Sirius T3 to measure log P values for 24 compounds of the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge set. For 13 of the selected compounds, experimental constraints set by solubility, lipophilicity, \(\hbox {p}{K}_{{\rm a}}\) properties of the analytes, and experiment analysis volume limitations of the Sirius T3 instrument resulted in an inability to achieve reliable log P measurements suitable for the blind challenge (Table S4). For example, SM24 has a basic \(\hbox {p}{K}_{{\rm a}}\) of 2.60 and we could not optimize log P measurement protocol because in the presence of octanol phase apparent \(\hbox {p}{K}_{{\rm a}}\) was shifting beyond the measurable pH range of the Sirius T3. On the other hand SM03 log P could not measured with potentiomentric method due to its low aqueous solubility. Only 11 of 24 compounds from the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge set were found to be suitable for potentiometric log P measurements with the Sirius T3. The resulting challenge dataset presented here has a log P range of 1.95–4.09. Six of these represent the 4-amino quinazoline scaffold (SM02, SM04, SM07, SM09, SM12, SM13). There are two benzimidazoles (SM14, SM15), one pyrazolo[3,4-d]pyrimidine (SM11), one pyridine (SM16), and one 2-oxoquinoline (SM08) (Fig. 3). The mean and SEM of replicate log P measurements, SAMPL6 compound IDs (SMXX), and SMILES identifiers of these compounds are presented in Table 1. In all cases, the SEM of the log P measurements ranged between 0.01–0.07 \(\log _{10}\) units.
Results of independent replicate measurements are presented in Table S2. Preparation of each replicate sample started from weighing dry powder of the same analyte lot. The log P estimates from potentiometric titrations were evaluated using the partitioning and ionization equilibrium model as implemented in the Sirius T3 Refine software, which produces log P estimates for both neutral and ionic species. We observed that log P values of neutral species were highly reproducible, while variance of log P of ionized species between replicate experiments was high. It was also not possible to measure log P values of the ionized species reliably as doing so would require sampling higher log R values. Since it was probihitively difficult to optimize experimental protocols to capture partitioning of ionic species accurately, we optimized the experiments to prioritize accurate measurement of neutral species log P (log P0) and constructed the blind computational prediction challenge based on log P0 values.

Illustrative potentiometric log P measurements of phenol (monoprotic, acid, log P 1.49) and SM16 (diprotic, amphoteric, log P 2.62) with the Sirius T3. Triangles represent experimental data points collected during the octanol-ISA water titrations and solid lines represent the ionization and partitioning model fit to the data. a, b Computed mean molecular charge vs pH. Mean molecular charge is calculated based on experimental \(\hbox {p}{K}_{{\rm a}}\) values and types (acid or base type) of the analyte. The black line is the model titration curve in aqueous media and based on the aqueous \(\hbox {p}{K}_{{\rm a}}\). Blue, red, and green triangles represent three sequential titrations with increasing log R (increasing octanol) that show shifted \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values. The inflection point of titration curves indicates the \(\hbox {p}{K}_{{\rm a}}\) or \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\), though these values are obtained by a global fit. For titration of acidic species, partitioning into the octanol phase increases the observed \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\). In the titration of the basic \(\hbox {p}{K}_{{\rm a}}\) of SM16, increasing log R causes a decrease in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\). The pH range of the experiment was determined such that only the titration of basic \(\hbox {p}{K}_{{\rm a}}\) was captured (molecular charge between +1 and 0). c, d log R profiles show a shift in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) with respect to increasing relative octanol volume. These plots aid in the design of the experiment and selection of optimal octanol volumes that aim to maximize separation between \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values for better model fit within experimental limitations (pH and analysis vial volume). e, f Buffer index profiles show buffering capacity observed in three titrations with increasing log R (blue to green). The black line is the intrinsic buffering capacity of water. For an accurate potentiometric measurement, buffering capacity signal of the analyte must be above the buffering capacity of water. As octanol volume increases, the concentration of the analyte in aqueous phase, and thus buffering capacity, decreases. g, h Predicted relative populations of ionization states in octanol and water phases as a function of pH, based on the equilibrium model fit to experimental data.

Molecules included in the SAMPL6 Part II log P challenge set. Reliable experimental potentiometric log P measurements were collected for the 11 molecules depicted here. Reported uncertainties are expressed as the standard error of the mean (SEM) of replicate measurements. Molecules are depicted using OpenEye OEDepict Tool [61]. Canonical isomeric SMILES strings of all compounds are given in Table 1, and replicate log P measurements can be found in Table S2
Discussion
Dynamic range of log P measurements and solubility limitations
We attempted to measure the log P for all 24 SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge compounds, but the Sirius T3 potentiometric log P measurement method was able to provide reliable measurements for only a subset of 11 molecules which were included in the blind challenge. We only included molecules that yielded reliable, precise log P measurements in the computational blind challenge.
A number of factors restricted the ability to perform reliable log P measurements and led to elimination of some compounds from the initial set of 24: low water solubility within the pH range of the titration, the limited volume capacity of the glass sample vial which limits the maximum achievable octanol:water ratio, the octanol-dependent \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values shifting outside the measurable pH range of 2–12 (especially high acidic \(\hbox {p}{K}_{{\rm a}}\)s and low basic \(\hbox {p}{K}_{{\rm a}}\)s). If an analyte does not suffer from the issues mentioned above, dynamic range of this log P measurement method is limited by smallest (related to dispensing accuracy and evaporation rate) and largest octanol volumes (related to analysis vial volume) that can be dispensed.
Optimizing experimental protocols for each compound
For the set of compounds in SAMPL6 Challenge, we observed that the Sirius T3 potentiometric log P measurement experiments were in practice very low throughput because of the necessary iterative protocol optimization for each compound. The parameters determining a potentiometric log P experiment are: mass of analyte, initial volume of ISA water, three target volumes of octanol for sequential titrations with increasing log R, and pH range of the pH titration. Factors that were considered in this optimization and limitations of choice are discussed below.
Optimizing the sequence of octanol–water volumetric ratios and range of pH titration
To obtain reliable and precise log P estimates from experimental data, it is recommended to fit the ionization and partitioning equilibrium model to at least three potentiometric titrations with well separated \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values (Fig. 2a, b). log P values can also be estimated from two potentiometric titrations, but not as accurately. \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values of sequential titrations need to be at least 0.3 \(\hbox {p}{K}_{{\rm a}}\) units separated from one another and from the aqueous \(\hbox {p}{K}_{{\rm a}}\). To achieve this, selecting an optimal set of octanol–water volumetric ratios is key.
It is logical to target the largest difference in octanol volumes, but the minimum volume of aqueous phase that provides enough depth for the pH probe (1.4 mL) and maximum analysis vial volume (3 mL) result in only 1.6 mL of available volume for the octanol phase, limiting the maximum octanol:water volume ratio R to \(\sim 1.1\). Typically, one would pick octanol volumes for each of three sequential titrations that maximize the difference in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) by maximizing the difference in log R values as much as possible considering the other experimental constraints. Simulated log R profiles based on predicted log P and experimental \(\hbox {p}{K}_{{\rm a}}\) values provide guidance in the selection of octanol volumes (Fig. 2c, d). These plots show how much |\(\hbox {p}{K}_{{\rm a}}\)-\(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)| difference can be gained with respect to a change in log R, based on the titration and ionization propensity of each molecule, but they are only as useful as the accuracy of log P prediction. For that reason, potentiometric log P measurements needs to be optimized with an iterative process where the first experimental protocol is designed using predicted log P and experimental \(\hbox {p}{K}_{{\rm a}}\) of the analyte. Based on the \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) shifts and quality of titration curves observed, a second experiment is designed to improve \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) shifts by adjusting the octanol volumes after consulting the log R profile and using the estimated log P from the previous experiment as a guide. Sometimes 3 or 4 iterations were necessary to reach an optimal protocol that results in a good fit between predicted and experimental titration curves and produces reproducible log P estimates. An example protocol optimization for SM02 guided by log R values is shown in Fig. 4.

Potentiometric log P protocol optimization of SM02 based on log R. Experimental results of initial trial (a) and optimized protocol (b) are shown for SM02. log R profile before optimization (a lower panel) shows insufficient apparent \(\hbox {p}{K}_{{\rm a}}\) shift due to poor choice of octanol–water volume ratios. This experiment led to log P and log P+1 estimates of 4.32 and 1.38. For a good measurement, triangles that indicate |\(\hbox {p}{K}_{{\rm a}}\)-\(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)| of each titration in log R profiles must fall on the slope region of the log R profile instead of the plateau region. Adjusting log R by decreasing octanol volumes in each titration led to a better experiment with distinct titration curves and well separated \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values (b). log P and log P+1 were measured as 4.10 and 1.32 with the optimized protocol. Once we achieved optimization of potentiometric log P protocol, triplicate measurements were collected using the same protocol
While maximizing the difference in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values from each other and from the aqueous \(\hbox {p}{K}_{{\rm a}}\) is desirable, sometimes it is necessary to reduce the octanol volume to limit the shift in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) so that it remains within a measurable range. This would be necessary when the aqueous \(\hbox {p}{K}_{{\rm a}}\) is a weak acid (\(\hbox {p}{K}_{{\rm a}}\)\(>9\)) or weak base (\(\hbox {p}{K}_{{\rm a}}\)<5), since the presence of the octanol phase causes \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) shifts towards higher and lower values, respectively, approaching the limit of the measurable pH range of the instrument. Measurable pH range is mainly limited by the acid and base strength of titration solutions against the increasing buffering capacity of water at pH values below 2 and above 12. It is also important to mention that even if the \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) value itself is within the stated measurement range of 2–12, if a large portion of the titration curve is beyond limits (i.e., saturation of fractional population on both sides of the \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)), then the experimental titration curve may not be fit to the model titration curve exactly and \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) cannot be determined as precisely. When the dynamic part of the titration curve (\(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) ± 2) shifts outside of the measureable pH range, it reduces the confidence in \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) estimates of the fit. Therefore, \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values should ideally be at least ± 1 unit, and preferably ± 2 units away from the limits of pH measurement with this instrument, which can be extended to pH 1 and 13 at most. For this reason, it is easier to optimize log P experiments for monoprotic molecules which have acidic \(\hbox {p}{K}_{{\rm a}}\)s between 3–10 and basic \(\hbox {p}{K}_{{\rm a}}\)s between 4–11. Some molecules in the SAMPL6 set which were not suitable for potentiometric log P measurements because of this criteria were: SM01, SM17, SM18, SM19, and SM24 (Table S4).
Sample preparation considerations and determination of appropriate starting concentration
Sample preparation starts with the weighing of solid powder material to analysis vials. How much analyte to use is another important decision that requires optimization. General guidance according to the Sirius T3 manual is to use 1–10 mg, and the aqueous phase volume is typically adjusted to the minimum volume (1.4–1.5 mL). The buffering capacity and compound solubility are the two factors that guide lower and higher limits of suitable analyte concentration. The Sirius T3 produces buffer index vs pH plots (Fig. 2e, d) which provide guidance on how much analyte is needed for sufficient potentiometric signal. To guide the first experiment, these plots can be simulated based on analyte mass, experimental \(\hbox {p}{K}_{{\rm a}}\), predicted log P, and selected octanol volumes. In further iterations of experiments, the buffer index profiles of the previous experiment guides the decisions about how to optimize the protocol. On the other hand, aqueous solubility limits the maximum concentration of the analyte in the aqueous phase. Moreover, since the experimental methodology depends on measuring the \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) shift during pH titrations as species partition into the nonaqueous phase, the analyte must stay in solution over the titrated pH range for the entire experiment, as the presence of an insoluble phase represents another reservoir for compound partitioning that would invalidate the coupled ionization-and-partitioning model used to compute the log P. The pH titration range is adjusted to capture a sufficient region below and above the \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) to ensure ionization states with lower solubility are also visited (neutral and zwitterionic states).
For these compounds resembling fragments of kinase inhibitors –the compounds considered in the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge [29] and this study– this solubility criterion turned out to be very challenging to meet. A large portion of compounds in the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge set were found to be insufficiently soluble for potentiometric log P experiments at some region of the pH range that needs to be titrated during the experiment, more likely the pH region where the neutral population of analytes are prominent. These compounds for which potentiometric log P measurement could not be optimized due to solubility limitations are listed in Table S4. For other compounds, we had to try reducing the analyte sample quantity from 3 mg to 1 mg of compound to find the optimum balance between ensuring the compound remained fully soluble and ensuring sufficiently high buffering capacity signal. The rate of change of pH vs. volume of acid or base titrated in analyte solution must differ from the rate of pH change in just water. This quantity is expressed as a buffering index in buffer index profiles generated by Sirius T3 (Fig. 2e, f), where a black solid line describes the theoretical buffering capacity of water and colored triangles describe the experimental buffering capacity of the analyte. For high quality measurements, reaching at least 0.001 buffer index at the maximum point of the titration (at pH that equals \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\)) is recommended.
In our case, the exact solubility of compounds was not known prior to log P measurements. We had to evaluate precipitation issues based on the distortion of mean molecular charge vs pH profiles (Fig. 2e, d) from ideal shape by adjusting starting analyte masses until the distortions disappear. Distortions manifest as very steep drops or oscillations in relative ionization state populations with respect to pH. An example is shown in Fig 5A Sample Ionization Graph. The turbidity indicator of Sirius T3 can not be utilized for solubility detection during log P experiments since the presence of octanol causes turbidity in the aqueous phase due to vigorous stirring during titrations. Predosing 80–100 μL octanol before addition of ISA water, as well as sonication and stirring after titrant addition, were also helpful for overcoming kinetic solubility problems. An example protocol optimization for SM08 to overcome solubility problems is shown in Fig 5.

Potentiometric log P protocol optimization for SM08 to alleviate aqueous solubility problems. Experimental results of initial protocol (a) and the optimized protocol (b) are shown for SM08. Both first (blue) and second (red) titrations in Sample Ionization Graph before optimization (a lower panel) show deviation from expected sigmoidal shape which is an indication of an insoluble analyte. This experiment with solubility issues led to log P and log P+1 estimates of 3.97 and 1.86. To eliminate precipitation, we could not lower analyte mass below 1 mg. Instead we were able to optimize the experimental protocol by increasing the predosed octanol volume and increasing additional octanol volumes added in each titration. Predosing octanol helps only with kinetic solubility issues. Larger octanol volumes can help to improve the experiment when thermodynamic solubility is the limitation, by allowing larger amounts of analyte partitioning into the octanol phase and reducing the analyte concentration in aqueous phase. Additional octanol volumes were selected such that they would also improve log R profile of the measurement. With optimized protocol (b) we achieved sample ionization profiles without any precipitation effects. log P and log P+1 were measured as 3.16 and 0.23. Once we achieved optimization of potentiometric log P protocol, triplicate measurements were collected using the same protocol
If possible, measuring solubility of compounds prior to potentiometric log P measurements can provide helpful information for more efficient log P measurement protocol optimization. However, since solubility is pH-dependent, the lowest solubility of the compound during the entire pH 2–12 range would be the information necessary to guide the experimental design. An experiment for a compound with 400 g/mol molecular weight using the minimum analysis made of 1 mg and 1.5 mL of aqueous phase corresponds to 1.67 mM. To be suitable for potentiometric log P measurements with the Sirius T3, at least 1.67 mM aqueous solubility is necessary throughout the pH range of the analysis.
One way to increase the dynamic range of potentiometric log P measurement with the Sirius T3 is to increase the range of log R that can be sampled by performing three different \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) measurements in three different analysis vials instead of three sequential titrations in one vial. But since log R is dependent on the cumulative octanol volume in sequential titrations, the advantage of the single titration approach is not significant. The single titration approach can only allow a small additional volume for octanol phase which would be used to dispense multiple acid and base stock solution volumes (\(\sim\)0.2 mL). We did not elect to investigate this design because we did not want to introduce another source of error: the variance in sample mass between measurements. Since the initial sample mass is an input parameter to the experimental model, using three different sample masses would introduce effects coming from the inaccuracy of the analytical balance to log P estimates.
Another way to prepare analyte samples for Sirius T3 measurements is to start from DMSO stock solutions instead of dry powder stocks. However, potentiometric measurements require 1–10 mg/mL analyte concentration in order to reach sufficient buffering capacity. The required concentration of the DMSO stock solution would be quite high, and sometimes impossible due to solubility limits in DMSO. Typical DMSO stock solution concentrations are 10 mM. For an analyte with 400 g/mol molecular weight, the concentration of 10 mM DMSO stock solution corresponds to 4 mg/mL. In order to achieve the minimum required 1 mg/mL analyte solution for the Sirius T3 experiment, the aqueous phase would have to consist of 25% DMSO which would cause significant cosolvent effects. On the other hand, achieving lower cosolvent presence, such as 2.5% DMSO, would require DMSO stock solutions of 100 mM at which concentration the analyte may not be soluble. Presence of cosolvent at even low amounts is undesirable due to the potential the effect on the log P measurements. Therefore, it is not recommended to perform these experiments starting from DMSO stock solutions.
Reliable determination of log P values of ionized species was not possible
Although it is possible to use Sirius T3 potentiometric log P measurements to determine the partition coefficients of ionic species as well, in practice, we were not able to achieve log P1 estimates with low variance between experiments. The partitioning of ionic species into the organic phase is typically much lower than that of the neutral species, and to capture this accurately by measuring sufficiently large \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) shifts, it would be necessary to use much larger octanol to water volumetric ratios R. The Sirius T3 glass analyte vials can hold up to 3 mL, which limits the maximum achievable octanol to water volumetric ratio. Since at least 1.4 mL must be devoted to the aqueous phase for the pH probe, this leaves only 1.6 mL for the octanol phase, producing a maximum achievable \(R \sim 1.1\). Another limitation was the measureable pH range. Since log P measurements rely on determining well-separated \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values at different log R values to get a good model fit, the octanol to water volumetric ratio needs to be selected such that \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values are well separated but not out of the measurable pH range (2–12).
To capture the partitioning of ionic species to the octanol layer reliably, experiments need to be set up with larger log R ratios which is problematic if this causes \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) to shift outside of the measureable pH range. Therefore, we designed the experiments to capture only the partition coefficient of the neutral species (log P0) accurately. The SAMPL6 log P Prediction Challenge was constructed only on prediction of neutral species.
The lack of reliable determination of partition coefficient values of the ionic species (log P+1 or log P−1) may be a source of systematic error in the estimate of log P of the neutral species (log P0). For hydrophobic compounds with negligible partitioning of the ionic species into the octanol-rich phase (log P+1, log P−1\(\le\) 2), log P0 estimates would still be accurate even if ion partitioning is not captured well. For compounds that may have higher levels of ionic partitioning, to minimize the impact of inaccurate log P+1 or log P−1 experimental estimates on log P0 measurements, we used ACD/Labs predicted log P+1 and log P−1 values as the starting point for the refinement of the ionization and partitioning equilibrium model parameters (performed with Sirius T3 Refine Software).
Absence of zwitterions allowed accurate log P measurements of amphoteric molecules
Multiple publications point out discrepancies between log P values determined by the potentiometric method and the shake-flask experiments for zwitterionic compounds [62, 63]. There are multiprotic compounds in the SAMPL6 dataset (SM14, SM15, and SM16), but we believe these measurements were not affected by this problem because they are not zwitterionic. Zwitterionic molecules have a zwitterion as the dominant neutral state in the pH region between the two \(\hbox {p}{K}_{{\rm a}}\)s (a lower acidic \(\hbox {p}{K}_{{\rm a}}\) and a higher basic \(\hbox {p}{K}_{{\rm a}}\)). SM14 has two basic \(\hbox {p}{K}_{{\rm a}}\)s and is not found as a zwitterion at any pH between 2 and 12. SM15 and SM16 are amphoteric compounds that possess both acidic and basic titratable groups, however, according to spectrophotometric \(\hbox {p}{K}_{{\rm a}}\) measurements in the presence of cosolvent their acidic \(\hbox {p}{K}_{{\rm a}}\) values are higher than their basic \(\hbox {p}{K}_{{\rm a}}\) values. This means the major neutral form of these compound is the non-charged state, not a zwitterion. Spectrophotometric \(\hbox {p}{K}_{{\rm a}}\) measurements with varying percentage of methanol as cosolvent were performed with the Sirius T3 and included in supplementary documents. Acidic or basic character of macroscopic \(\hbox {p}{K}_{{\rm a}}\) values was assigned based on the slope of Yasuda-Shedlovsky plots.
In addition, quantum mechanics calculations [64] do not predict the presence of multiple tautomers of the neutral state at significant populations for any of the molecules in the SAMPL6 log P challenge set. Possible tautomers, such as the zwitterionic state, are predicted to be much higher in energy and thus unlikely to play a significant role even if we considered a prediction error margin for quantum mechanics-based calculations. Therefore, we do not think our potentiometric log P measurements are influenced by presence of zwitterions or minor tautomeric forms.
Suggestions for future log P data collection
High quality datasets of experimental physicochemical property measurements are valuable for testing computational predictions. Benchmarking and evaluation efforts like the SAMPL challenges benefit from large experimental datasets with diverse chemical species. The quality of log P measurements collected with the Sirius T3 potentiometric method are satisfactory and comparable to gold standard shake flask measurements [45, 49, 51]. The Sirius T3 potentiometric log P method requires aqueous \(\hbox {p}{K}_{{\rm a}}\)s to be measured experimentally ahead of time. The ability to obtain log P measurements of neutral and charged species separately, instead of measuring pH dependent log D, is a unique advantage of the Sirius T3 approach compared to shake-flask or HPLC-based methods where ionization effects are involved with partitioning behaviour. However, due to previously discussed limitations and the necessity for extensive protocol optimization for each analyte, we are reluctant to suggest potentiometric log P measurements with the Sirius T3 as a general and high-throughput method for future log P data collection unless significant resources and work hours of a human expert can be dedicated to protocol optimization and data collection.
Informed selection of analytes can help improve the success of Sirius T3 experiments. For example, this approach is easier to apply to highly soluble compounds (more than 1 mg/mL solubulity in 0.15 M KCl through the entire range of pH titration range at room temperature) with \(\hbox {p}{K}_{{\rm a}}\) values in the midrange (3<acidic \(\hbox {p}{K}_{{\rm a}}\)<10 and 4<basic \(\hbox {p}{K}_{{\rm a}}\)<11). There is no significant difference in difficulty between the measurements of monoprotic vs multiprotic compounds, as long as one of the \(\hbox {p}{K}_{{\rm a}}\) values of the multiprotic compound is in the midrange. For determining the log P of neutral species, it is sufficient to collect potentiometric titration data between the neutral state and the + 1 or − 1 charged states by titrating the pH region that captures the relevant \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) values. It is not necessary to capture the titration of a second \(\hbox {p}{K}_{{\rm a}}\) (Fig. 2b).
Our opinion is that log D measurements at a buffered pH can be much more easily obtained in a higher throughput fashion using miniaturized shake-flask measurements, such as those used in SAMPL5 log D Challenge experimental data collection [21]. To obtain log P values from experiments that were designed to measure log Ds, it is necessary to measure the \(\hbox {p}{K}_{{\rm a}}\) of compounds (such as with the Sirius T3) and conduct log D measurements using a buffered aqueous phase at a pH that will ensure that the analyte is completely in the neutral state. According to our experience, optimizing \(\hbox {p}{K}_{{\rm a}}\) measurements with the Sirius T3 is significantly easier than optimizing log P measurements, especially if a spectrophotometric (UV-metric) \(\hbox {p}{K}_{{\rm a}}\) method can be used instead of potentiometric, which is not an option for log P measurements.
Conclusion
This study reports the collection of experimental data for the SAMPL6 Part II log P Blind Prediction Challenge. In the physicochemical property prediction challenge components of SAMPL6, we aimed to separately evaluate performance of computational methods for predicting ionization (\(\hbox {p}{K}_{{\rm a}}\)) and nonaqeuous partitioning (log P) of small molecules, collecting experimental data for these properties on the same set of compounds and fielding sequential, independent prediction challenges. While we attempted to measure octanol–water log P for all compounds in the SAMPL6 \(\hbox {p}{K}_{{\rm a}}\) Challenge set—consisting of 24 compounds that resemble fragments of kinase inhibitors—experimental limitations of the Sirius T3 potentiometric log P method meant that reliable log P measurements could only be performed for 11 of these compounds. The resulting compound set had meaured log P values in the range of 1.95–4.09. This set included six molecules with 4-aminoquinazoline scaffolds, and two molecules with benzimidazole scaffolds. Although the chemical diversity and number of compounds was rather limited, blind high-quality log P datasets are rare, and still highly valuable for evaluating the performance of computational predictions. Therefore, the SAMPL6 Part II log P Blind Prediction Challenge was held between November 1, 2018 and March 22 ,2019 using the log P measurements presented in this paper. This dataset can be utilized as part of a benchmark set for the assessment of future log P predictions methods.
Data availability statement
All SAMPL6 log P Challenge instructions, submissions, experimental data and analysis are available at https://github.com/samplchallenges/SAMPL6/tree/master/physical_properties/logP
Notes
SAMPL6 was originally announced as featuring a log D prediction challenge, but there were difficulties in the collection of experimental data. The original plan was to measure log P0, log P−1, and log P+1 and calculate log D values at the experimental pH using these values. However, we were able to measure the partition coefficients of neutral species (log P0) much more reliably than ionic species with potentiometric log P method of Sirius T3, as elaborated further below.
Abbreviations
- SAMPL:
-
Statistical Assessment of the Modeling of Proteins and Ligands
- log P :
-
log\(_{10}\) of the organic solvent-water partition coefficient (\(K_{ow}\), refers to partition of neutral species unless stated otherwise)
- log D :
-
log\(_{10}\) of organic solvent-water distribution coefficient (\(D_{ow}\))
- log R :
-
log\(_{10}\) of the volumetric ratios of partition solvents (octanol to water)
- \(\hbox {p}{K}_{{\rm a}}\) :
-
−log\(_{10}\) of the acid dissociation equilibrium constant
- \(\hbox {p}_{{\rm o}}{K}_{{\rm a}}\) :
-
−log\(_{10}\) apparent acid dissociation equilibrium constant in octanol–water biphasic system
- ISA:
-
Ionic-strength adjusted solution with 0.15 M KCl
- SEM:
-
Standard error of the mean
- LC-MS:
-
Liquid chromatography-mass spectrometry
- NMR:
-
Nuclear magnetic resonance spectroscopy
- HRMS:
-
High-resolution mass spectrometry
- octanol:
-
1-Octanol, also known as n-octanol
References
Mobley DL, Chodera JD, Isaacs L, Gibb BC (2016) Advancing predictive modeling through focused development of model systems to drive new modeling innovations. UC Irvine: Department of Pharmaceutical Sciences, UCI. https://escholarship.org/uc/item/7cf8c6cr
Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD, Pande VS (2008) Predicting small-molecule solvation free energies: an informal blind test for computational chemistry. J Med Chem 51(4):769–779. https://doi.org/10.1021/jm070549+
Guthrie JP (2009) A blind challenge for computational solvation free energies: introduction and overview. J Phys Chem B 113(14):4501–4507. https://doi.org/10.1021/jp806724u
Skillman AG, Geballe MT, Nicholls A (2010) SAMPL2 challenge: prediction of solvation energies and tautomer ratios. J Comput Aided Mol Des 24(4):257–258. https://doi.org/10.1007/s10822-010-9358-0
Geballe MT, Skillman AG, Nicholls A, Guthrie JP, Taylor PJ (2010) The SAMPL2 blind prediction challenge: introduction and overview. J Comput Aided Mol Des 24(4):259–279. https://doi.org/10.1007/s10822-010-9350-8
Skillman AG (2012) SAMPL3: blinded prediction of host–guest binding affinities, hydration free energies, and trypsin inhibitors. J Comput Aided Mol Des 26(5):473–474. https://doi.org/10.1007/s10822-012-9580-z
Geballe MT, Guthrie JP (2012) The SAMPL3 blind prediction challenge: transfer energy overview. J Comput Aided Mol Des 26(5):489–496. https://doi.org/10.1007/s10822-012-9568-8
Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK (2012) Blind prediction of host-guest binding affinities: a new SAMPL3 challenge. J Comput Aided Mol Des 26(5):475–487. https://doi.org/10.1007/s10822-012-9554-1
Guthrie JP (2014) SAMPL4, a blind challenge for computational solvation free energies: the compounds considered. J Comput Aided Mol Des 28(3):151–168. https://doi.org/10.1007/s10822-014-9738-y
Mobley DL, Wymer KL, Lim NM, Guthrie JP (2014) Blind prediction of solvation free energies from the SAMPL4 challenge. J Comput Aided Mol Des 28(3):135–150. https://doi.org/10.1007/s10822-014-9718-2
Muddana HS, Fenley AT, Mobley DL, Gilson MK (2014) The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des 28(4):305–317. https://doi.org/10.1007/s10822-014-9735-1
Mobley DL, Liu S, Lim NM, Wymer KL, Perryman AL, Forli S, Deng N, Su J, Branson K, Olson AJ (2014) Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aided Mol Des 28(4):327–345. https://doi.org/10.1007/s10822-014-9723-5
Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK (2017) Overview of the SAMPL5 host-guest challenge: are we doing better? J Comput Aided Mol Des 31(1):1–19. https://doi.org/10.1007/s10822-016-9974-4
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) Blind prediction of cyclohexane–water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des 30(11):1–18. https://doi.org/10.1007/s10822-016-9954-8
Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL et al (2018) Overview of the SAMPL6 host-guest binding affinity prediction challenge. J Comput Aided Mol Des 32(10):937–963
Muddana HS, Gilson MK (2012) Prediction of SAMPL3 host-guest binding affinities: evaluating the accuracy of generalized force-fields. J Comput Aided Mol Des 26(5):517–525. https://doi.org/10.1007/s10822-012-9544-3
Gibb CLD, Gibb BC (2013) Binding of cyclic carboxylates to octa-acid deep-cavity cavitand. J Comput Aided Mol Des 28(4):319–325. https://doi.org/10.1007/s10822-013-9690-2
Cao L, Isaacs L (2014) Absolute and relative binding affinity of cucurbit[7]uril towards a series of cationic guests. Supramol Chem 26(3–4):251–258. https://doi.org/10.1080/10610278.2013.852674
Sullivan MR, Sokkalingam P, Nguyen T, Donahue JP, Gibb BC (2017) Binding of carboxylate and trimethylammonium salts to octa-acid and TEMOA deep-cavity cavitands. J Comput Aided Mol Des 31(1):1–8. https://doi.org/10.1007/s10822-016-9925-0
Bannan CC, Burley KH, Chiu M, Shirts MR, Gilson MK, Mobley DL (2016) Blind prediction of cyclohexane-water distribution coefficients from the SAMPL5 challenge. J Comput Aided Mol Des 30(11):927–944. https://doi.org/10.1007/s10822-016-9954-8
Rustenburg AS, Dancer J, Lin B, Feng JA, Ortwine DF, Mobley DL, Chodera JD (2016) Measuring experimental cyclohexane-water distribution coefficients for the SAMPL5 challenge. J Comput Aided Mol Des 30(11):945–958. https://doi.org/10.1007/s10822-016-9971-7
Waring MJ (2010) Lipophilicity in drug discovery. Expert Opin Drug Discov 5(3):235–248. https://doi.org/10.1517/17460441003605098
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23(1):3–25. https://doi.org/10.1016/S0169-409X(96)00423-1
Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71(6):525–616. https://doi.org/10.1021/cr60274a001
Arnott JA, Planey SL (2012) The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov 7(10):863–875. https://doi.org/10.1517/17460441.2012.714363
Lipnick RL (2008) Environmental hazard assessment using lipophilicity data. In: Pliška V, Testa B, van de Waterbeemd H (eds) Methods and principles in medicinal chemistry. Wiley-VCH Verlag GmbH, Weinheim, pp 339–353. https://doi.org/10.1002/9783527614998.ch19
Pickard FC, König G, Tofoleanu F, Lee J, Simmonett AC, Shao Y, Ponder JW, Brooks BR (2016) Blind prediction of distribution in the SAMPL5 challenge with QM based protomer and pKa corrections. J Comput Aided Mol Des 30(11):1–14. https://doi.org/10.1007/s10822-016-9955-7
Işık M, Bergazin TD, Fox T, Rizzi A, Chodera JD, Mobley DL (2019) Assessing the accuracy of octanol-water partition coefficient predictions in the SAMPL6 Part II logP challenge. J Comput Aided Mol Des (SAMPL6 Part II special issue)
Işık M, Levorse D, Rustenburg AS, Ndukwe IE, Wang H, Wang X, Reibarkh M, Martin GE, Makarov AA, Mobley DL, Rhodes T, Chodera JD (2018) pKa measurements for the SAMPL6 prediction challenge for a set of kinase inhibitor-like fragments. J Comput Aided Mol Des 32(10):1117–1138. https://doi.org/10.1007/s10822-018-0168-0
Tsuji A, Kubo O, Miyamoto E, Yamana T (1977) Physicochemical properties of \(\beta\)-lactam antibiotics: oil-water distribution. J Pharm Sci 66(12):1675–1679. https://doi.org/10.1002/jps.2600661205
Avdeef A, Box KJ, Comer JEA, Hibbert C, Tam KY (1998) pH-Metric logP 10. Determination of liposomal membrane-water partition coefficients of lonizable drugs. Pharm Res 15(2):209–215. https://doi.org/10.1023/A:1011954332221
Octanol-Water Partitioning (2012) In: Absorption and drug development. Wiley, Hoboken pp 174–219. https://doi.org/10.1002/9781118286067.ch4
OECD (1995) Test No. 107: partition coefficient (n-octanol/water): shake flask method, OECD guidelines for the testing of chemicals, section 1. OECD Publishing, Paris. https://doi.org/10.1787/9789264069626-en
Hitzel L, Watt AP, Locker KL (2000) An increased throughput method for the determination of partition coefficients. Pharm Res 17(11):1389–1395. https://doi.org/10.1023/A:1007546905874
Lin B, Pease JH (2013) A novel method for high throughput lipophilicity determination by microscale shake flask and liquid chromatography tandem mass spectrometry. Comb Chem High Throughput Screen 16(10):817–825. https://doi.org/10.2174/1386207311301010007
De Bruijn J, Busser F, Seinen W, Hermens J (1989) Determination of octanol/water partition coefficients for hydrophobic organic chemicals with the “slow-stirring” method. Environ Toxicol Chem 8(6):499–512. https://doi.org/10.1002/etc.5620080607
Andersson JT, Schräder W (1999) A method for measuring 1-octanol water partition coefficients. Anal Chem 71(16):3610–3614. https://doi.org/10.1021/ac9902291
Herbert BJ, Dorsey JG (1995) n-Octanol-water partition coefficient estimation by micellar electrokinetic capillary chromatography. Anal Chem 67(4):744–749. https://doi.org/10.1021/ac00100a009
Berthod A, Carda-Broch S (2004) Determination of liquid-liquid partition coefficients by separation methods. J Chromatogr A 1037(1–2):3–14. https://doi.org/10.1016/j.chroma.2004.01.001
McCall JM (1975) Liquid-liquid partition coefficients by high-pressure liquid chromatography. J Med Chem 18(6):549–552. https://doi.org/10.1021/jm00240a003
Unger SH, Cook JR, Hollenberg JS (1978) Simple procedure for determining octanol aqueous partition, distribution, and ionization coefficients by reversed-phase high-pressure liquid chromatography. J Pharm Sci 67(10):1364–1367. https://doi.org/10.1002/jps.2600671008
Valkó K, Bevan C, Reynolds D (1997) Chromatographic hydrophobicity index by fast-gradient RP-HPLC: a high-throughput alternative to log P/log D. Anal Chem 69(11):2022–2029. https://doi.org/10.1021/ac961242d
Valkó K (1984) General approach for the estimation of octanol/water partition coefficient by reversed-phase high-performance liquid chromatography. J Liq Chromatogr 7(7):1405–1424. https://doi.org/10.1080/01483918408074054
OECD (2004) Test No. 117: partition coefficient (n-octanol/water): HPLC method, OECD guidelines for the testing of chemicals, section 1. OECD Publishing, Paris. https://doi.org/10.1787/9789264069824-en
Takacs-Novak K, Avdeef A (1996) Interlaboratory study of log P determination by shake-flask and potentiometric methods. J Pharm Biomed Anal 14:1405–1413
Avdeef A (1992) PH-metric log P. Part 1. Difference plots for determining ion-pair octanol-water partition coefficients of multiprotic substances. Quant Struct Act Relatsh 11(4):510–517
Sirius T3 User Manual, v1.1 (2008) Sirius Analytical Instruments Ltd. East Sussex, UK
Avdeef A (1993) pH-metric log P. II: refinement of partition coefficients and lonization constants of multiprotic substances. J Pharm Sci 82(2):183–190
Slater B, McCormack A, Avdeef A, Comer JE (1994) Ph-metric log P. 4. Comparison of partition coefficients determined by HPLC and potentiometric methods to literature values. J Pharm Sci 83(9):1280–1283
Comer J, Tam K (2001) Lipophilicity profiles: theory and measurement. Wiley-VCH, Zürich
OECD (2000) Guideline 122: partition coefficient (n-octanol/Water), pH-metric method for ionisable substances. Revised Draft
Işık M, Rustenburg A, Rizzi A, Bannan C, Gunner MR, Mobley DL, Chodera JD (2019) Accuracy of macroscopic and microscopic pKa predictions of small molecules evaluated by the SAMPL6 blind prediction challenge. J Comput Aided Mol Des. Manuscript in preparation
Wishart DS (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34(90001):D668–D672. https://doi.org/10.1093/nar/gkj067
Pence HE, Williams A (2010) ChemSpider: an online chemical information resource. J Chem Educ 87(11):1123–1124. https://doi.org/10.1021/ed100697w
NCI Open Database, August 2006 Release. https://cactus.nci.nih.gov/download/nci/
Enhanced NCI Database Browser 2.2. https://cactus.nci.nih.gov/ncidb2.2/
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH (2016) PubChem substance and compound databases. Nucleic Acids Res 44(D1):D1202–D1213. https://doi.org/10.1093/nar/gkv951
Tam KY, Takács-Novák K (2001 Apr) Multi-wavelength spectrophotometric determination of acid dissociation constants: a validation study. Anal Chim Acta 434(1):157–167. https://doi.org/10.1016/S0003-2670(01)00810-8
Allen RI, Box KJ, Comer JEA, Peake C, Tam KY (1998) Multiwavelength spectrophotometric determination of acid dissociation constants of ionizable drugs. J Pharm Biomed Anal 17(4–5):699–712. https://doi.org/10.1016/S0731-7085(98)00010-7
Ahn S, Fessler JA (2003) Standard errors of mean, variance, and standard deviation estimators. EECS Department, The University of Michigan. pp 1–2. http://web.eecs.umich.edu/~fessler/papers/lists/files/tr/stderr.pdf
OEDepict Toolkit 2017. Feb. 1. OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com
Takács-Novák K, Avdeef A, Box KJ, Podányi B, Szász G (1994) Determination of protonation macro- and microconstants and octanol/water partition coefficient of the antiinflammatory drug niflumic acid. J Pharm Biomed Anal 12(11):1369–1377. https://doi.org/10.1016/0731-7085(94)00090-5
Port A, Bordas M, Enrech R, Pascual R, Rosés M, Ràfols C, Subirats X, Bosch E (2018) Critical comparison of shake-flask, potentiometric and chromatographic methods for lipophilicity evaluation (Log P o/w ) of neutral, acidic, basic, amphoteric, and zwitterionic drugs. Eur J Pharm Sci 122:331–340. https://doi.org/10.1016/j.ejps.2018.07.010
Tielker N, Tomazic D, Eberlein L, Güssregen S, Kast SM (2019) The SAMPL6 challenge on predicting octanol-water partition coefficients from ECRISM theory. J Comput Aided Mol Des (SAMPL6 Part II special issue)
Acknowledgements
MI and JDC acknowledge support from the Sloan Kettering Institute. JDC acknowledges partial support from NIH Grant P30 CA008748. MI, JDC, ASR, and DLM gratefully acknowledge support from NIH Grant R01GM124270 supporting the SAMPL Blind Challenges. MI acknowledges support from a Doris J. Hutchinson Fellowship. DLM appreciates financial support from the National Institutes of Health (1R01GM108889-01), the National Science Foundation (CHE 1352608). The authors are extremely grateful for the assistance and support from the MRL Preformulations and NMR Structure Elucidation groups for materials, expertise, and instrument time, without which this SAMPL challenge would not have been possible. The authors would like to thank Ryan Cohen from the NMR Structure Elucidation group for the NMR and LC-MS analysis of SM13. MI and DL are grateful to Pion/Sirius Analytical for their technical support in the planning and execution of this study. We are especially thankful to Karl Box (Sirius Analytical) for the guidance on optimization and interpretation of log P measurements with the Sirius T3. We thank Brad Sherborne (MRL; ORCID: 0000-0002-0037-3427) for his valuable insights at the conception of the log P challenge and connecting us with TR and DL who were able to provide resources for experimental measurements. We acknowledge contributions from Caitlin Bannan who provided feedback on experimental data collection and structure of log P challenge from a computational chemist’s perspective. MI and JDC are grateful to OpenEye Scientific for providing a free academic software license for use in this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank anonymous reviewers for their input and constructive comments that improved this manuscript. Research reported in this publication was supported by National Institute for General Medical Sciences of the National Institutes of Health under Award Number R01GM124270 and R01GM108889, and from the National Cancer Institute of the National Institutes of Health under P30CA008748.
Disclaimers
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conceptualization, MI, JDC, TR, DLM ; Methodology, MI, DL; Software, MI ; Formal Analysis, MI ; Investigation, MI, DL; Resources, TR, DL; Data Curation, MI ; Writing-Original Draft, MI, DL; Writing - Review and Editing, MI, DL, JDC, TR, DLM; Visualization, MI ; Supervision, JDC, TR, DLM ; Project Administration, MI ; Funding Acquisition, DLM, JDC, TR, MI.
Corresponding authors
Ethics declarations
Conflict of interest
JDC was a member of the Scientific Advisory Board for Schrödinger, LLC during part of this study. JDC and DLM are current members of the Scientific Advisory Board of OpenEye Scientific Software. The Chodera laboratory receives or has received funding from multiple sources, including the National Institutes of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biosciences, XtalPi, the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, The Einstein Foundation, and the Sloan Kettering Institute. A complete list of funding can be found at http://choderalab.org/funding.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Işık, M., Levorse, D., Mobley, D.L. et al. Octanol–water partition coefficient measurements for the SAMPL6 blind prediction challenge. J Comput Aided Mol Des 34, 405–420 (2020). https://doi.org/10.1007/s10822-019-00271-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-019-00271-3