
Abstract
The computational prediction of protein–ligand binding affinities is of central interest in early-stage drug-discovery, and there is a widely recognized need for improved methods. Low molecular weight receptors and their ligands—i.e., host–guest systems—represent valuable test-beds for such affinity prediction methods, because their small size makes for fast calculations and relatively facile numerical convergence. The SAMPL3 community exercise included the first ever blind prediction challenge for host–guest binding affinities, through the incorporation of 11 new host–guest complexes. Ten participating research groups addressed this challenge with a variety of approaches. Statistical assessment indicates that, although most methods performed well at predicting some general trends in binding affinity, overall accuracy was not high, as all the methods suffered from either poor correlation or high RMS errors or both. There was no clear advantage in using explicit versus implicit solvent models, any particular force field, or any particular approach to conformational sampling. In a few cases, predictions using very similar energy models but different sampling and/or free-energy methods resulted in significantly different results. The protonation states of one host and some guest molecules emerged as key uncertainties beyond the choice of computational approach. The present results have implications for methods development and future blind prediction exercises.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The ability to accurately predict the binding affinity of a ligand to a target protein could dramatically reduce the burden of chemical synthesis and experimental affinity assays to a more manageable number of compounds and, more importantly, accelerate the discovery of needed medications. While computational screening methods such as docking and empirical scoring methods are routinely used in lead identification and enrichment [1], there is still a need for higher-level computational methods able to reliably rank candidate ligands by affinity; i.e., by their binding free energies [2, 3]. Such methods would be valuable in a secondary, higher-precision stage of virtual screening, as well as in the computer-guided optimization of lead compounds.
One of the central challenges in developing such methods is validation. For one thing, testing detailed computational methods on protein–ligand systems can be quite time-consuming. Furthermore, because proteins are large and flexible, it is difficult to be sure that a calculation has sampled all thermodynamically relevant conformations; i.e., that the calculation has converged. One valuable alternative is to test whether the method can accurately reproduce experimental solvation free energies of small molecules, as discussed elsewhere in this issue. However, solvation free energies can be measured only for molecules that are volatile enough to be detectible in the gas phase, and such molecules tend to be relatively small and nonpolar. As a consequence, solvation free energies cannot be used to test how well a solvation model handles water in the complex microenvironment of a binding pocket. They also do not provide strong tests of the ability of a model to capture interactions between solutes in the aqueous environment.
Host–guest systems are attractive, though largely untapped, test cases for detailed models of binding. Although host molecules are not proteins, molecular recognition events in host–guest and protein–ligand systems are governed by the same statistical mechanical principles and driven by the same interaction forces [4]. In particular, since the binding pockets of hosts are well solvated, the intricacies of hydration can play an equally important role in determining their binding properties. Nonetheless, host molecules are dramatically simpler than proteins, because they typically comprise only a few hundred atoms and tend to be more rigid than proteins. As a consequence, it is dramatically easier to achieve adequate conformational sampling for them compared to proteins [5, 6]. In recent years, cucurbiturils have emerged as particularly appealing test cases, because they exhibit a wide range of affinities for varied guests in aqueous solution [7–9], with maximal affinities rivaling those of the tightest-binding protein–ligand systems [10–12]. In addition, a growing repertoire of cucurbituril derivatives offers the possibility of further diversity in terms of chemistries, molecular flexibility, etc. [13–16] The growing interest in these host–guest systems for applications in drug delivery [17, 18] and chemical sensors [19] is also generating rapid growth in the volume of published binding affinity data.
In addition to the selection of suitable test systems, the format of a validation study is of critical importance. In particular, efforts such as CASP [20], the pKa Cooperative [21], and SAMPL [22] have proven the value of blinded prediction challenges for objective validation of computational methods. Here we summarize the procedure and results of the first blinded prediction challenge for host–guest binding affinities, which was included as a new component of this year’s SAMPL. Other papers in this issue provide detailed reports from the participating groups. This host–guest affinity challenge bridges the scale and complexity of the other SAMPL prediction challenges, including prediction of tautomers, solvation free energies, and protein–ligand affinities. The primary goals of this challenge are to define the state-of-the-art, learn from each other’s experiences, identify problem areas requiring further improvement, create improved methods of modeling host–guest binding, and gain knowledge that will advance methods of predicting protein–ligand affinities.
Materials and methods
Challenge design
Information about the challenge was posted on the SAMPL website, sent to many commercial, industry, and academic researchers, and posted on the computational chemistry list (CCL; http://www.ccl.net). All interested parties, not only those to whom announcements were sent, were welcomed to take part by registering at the website (http://sampl.eyesopen.com). The data sets were provided for download there in SDF or PDB format. Participants were explicitly informed that the conformation, tautomer, and protonation states provided were not necessarily optimal or complete. Experimental conditions were specified, including the temperature, salt concentration, and pH at which the binding affinities were determined. In addition, a previous study by Ma et al. [15] was brought to the attention of all participants to ensure that every one was aware of relevant published data for host H1. The host H1 system was posted on the website on September 23rd, 2010, and the hosts H2 and H3 systems were posted on February 20th, 2010. Unfortunately, some participants were unaware that there were two sets of data and therefore submitted predictions for only host H1 or only hosts H2 and H3; a few did submit predictions for all three systems. The final submission deadline was June 30th, 2011. As in previous SAMPL challenges, the experimental values were not released to the participants until after the submission deadline. The outcome of the challenge and its implications were discussed at a workshop on August 1–2, 2011 at Stanford University, and all participants were invited to submit papers describing their calculations. The manuscripts, which were all reviewed by other SAMPL participants, are also published in this special journal issue.
Host–guest systems and structures
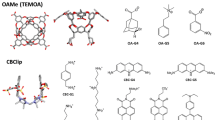
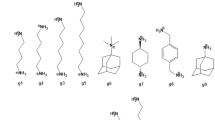
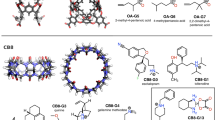
Crystallographic structures of hosts H1, H2 and H3 were obtained from the literature [15] or Cambridge structure database [23], and are shown in Fig. 1, along with the chemical structures of the guest molecules, G1–G9. These crystal structures of the hosts were provided to the participants along with modeled 3D coordinates of the guest molecules. Identifying the conformations, protonation state, and tautomer state of the hosts, guests and complexes, in solution was considered as part of the prediction challenge, and the modeled coordinates of the guest molecules were provided to the participants with the disclaimer that these are only meant to serve as potential starting geometries. The identifiers used here for some of the guests differ from the potentially ambiguous names used when originally identifying the compounds to the SAMPL3 participants. The original notation was: h1.guest(1–7) for guests G1–G7, h2.guest1/h3.guest1 for guest G8, and h2.guest2/h3.guest2 for guest G9. Note that other papers in this special issue may use the original notation to identify the guest molecules.

Crystal structures of hosts H1, H2, and H3, and chemical structures of guests G1–G9. Host H1 and its respective guests (G1–G7) are shaded in blue; hosts H2 and H3 with their respective common guests (G8 and G9) are shaded in orange
Experimental details
Drs. Lyle Isaacs (host H1 [15]) and Adam Urbach (hosts H2, H3 [24]) provided the unpublished experimental binding affinities of 11 new host–guest complexes. The data are summarized in Table 1. Binding affinities of the H1 systems were measured at 298 K in 20 mM sodium phosphate buffer at pH 7.4, in one or more of the following ways: (1) 1H NMR spectroscopy—titration of a fixed concentration of the guest with an increasing concentration of the host; (2) UV/Vis competition assay—titration of a fixed concentration of the host and a dye with an increasing concentration of guest; (3) 1H NMR competition assay. The binding affinity reported for G1 is that of a racemic mixture. A detailed description of the experimental protocols applied to similar host–guest systems can be found in Ref. [7]. Binding affinities of G8 and G9 with hosts H2 and H3 were determined using isothermal titration calorimetry (ITC) at 300 K in 10 mM sodium phosphate buffer at pH 7.0. Synthesis and characterization of compounds G8 and G9 will be reported elsewhere. Titration experiments were carried out on a VP-ITC calorimeter (Microcal, Inc.). The concentration of the host in the sample cell was between 0.062 and 0.174 mM, and the guest concentration was between 0.5 and 2.81 mM. The titration consisted of 27 consecutive injections of 10 μL with at least 200 s interval between injections. Heats of dilution, measured by titrating beyond saturation, were subtracted from each data set. All solutions were degassed prior to titration. The data were analyzed using the one-set-of-sites model in the Origin software; this is a simple binary equilibrium model that assumes no interaction between complexes. A high affinity of these guests (G8 and G9) with hosts H2 and H3 suggests inclusion complexation [25]. In all cases (hosts H1, H2, and H3), a 1:1 stoichiometry of binding was observed.
Computational methods
Participants employed a range of different methods, as detailed in other articles in this issue. Table 2 lists the methods by name, and categorizes them as follows:
-
Overall method
-
Pathway methods [2] compute binding free energies in terms of the free energy changes associated with a series of small steps along a path, such as a path linking the free and bound states. The pathway approaches used here include thermodynamic integration (TI) [26, 27], Bennett acceptance ratio (BAR) [28], Wang-Landau [29], binding energy distribution analysis method (BEDAM) [30], and orthogonal space random walk (OSRW) [31].
-
End-point methods compute binding free energies through analysis of only the free and bound states of the system, rather than connecting them by a path. The end-point methods used here include MM/PBSA [32], MM/GBSA [32], solvated interaction energy (SIE) [33], and Mining Minima (M2) [34, 35].
-
-
Potential energy as a function of conformation
-
Most of the methods used a generalized force field, OPLS [36, 37], GAFF [38], MMFF94 [39], CHARMm [40], or CGenFF [41], as an efficient method of estimating the potential energy of a molecule or complex as a function of conformation. Partial atomic charges, which are a critical force field component, were in turn generated by several different approaches [42–44], but most commonly AM1-BCC [45, 46].
-
One participant avoided the need to generate force field parameters, with their potentially non-optimal parameters and simplified functional form, by using an electronic structure method, density functional theory [47, 48] with the B3LYP functional [49, 50].
-
-
Solvation free energy as a function of conformation
-
All end-point methods used an implicit solvent model based on continuum electrostatics, such as PBSA [51], GBSA [52], PCM [53], and FiSH [54]. In addition, the BEDAM pathway method used the similar but more intricate AGBNP2 model [55, 56]. Note, however, that different implementations of nominally similar continuum electrostatic models might yield significantly different results if they use different atomic radii, and the use of different numerical solvers may also be relevant; in particular, the SIE method uses a boundary element solver instead of the more commonly used finite difference solver.
-
All pathway methods other than BEDAM used the TIP3P [57] explicit water model in the context of molecular dynamics (MD) simulations.
-
-
Conformational sampling method
-
All pathway methods used MD as the conformational sampling method.
-
Several end-point methods, generated and processed conformations by using a docking program to fit the guests into the crystal structures of their respective hosts.
-
The MM/GBSA and MM/PBSA methods sampled multiple conformations from the Boltzmann distribution with MD using explicit solvent.
-
The M2 method used Tork [58], an aggressive conformational search algorithm, along with an implicit solvent model, to identify many local energy minima for the free and bound structures.
-
-
Configurational entropy; i.e., the entropy associated with the host and guest, but not the solvent
-
The SIE method did not use any specific method to account for changes in configurational entropy on binding, although the <1 scaling parameter applied to the overall binding energy might be considered to account crudely for an entropic penalty proportional to energy.
-
The pathway methods include entropy implicitly but, in principle, fully, through their free energy approaches.
-
A number of end-point methods used the rigid-rotor/harmonic oscillator (RRHO) approximation.
-
The M2 method applied the harmonic approximation with mode scanning (HA/MS) to each energy well, and also accounted for the entropy associated with the existence of multiple energy wells. The HA/MS method is similar to RRHO, but it accounts for some anharmonicity in each energy well by scanning energies along eigenvectors of the Hessian associated with low eigenvalues (force constants).
-
Results
Overview of host–guest predictions
A total of 19 submissions were provided by 10 different research groups. Scatter plots of calculated versus experimental binding free energies are shown in Fig. 2, and statistical measures of accuracy are provided in the last two columns of Table 2. Because not all groups made predictions for all systems, and because the host H1 systems are quite different physically from the host H2 and host H3 cases, we present the error statistics separately for these systems in Table 2, under the headings Host1 and Host2/Host3. For those methods which were applied to all three hosts, error statistics across all three hosts are also provided at the bottom of Table 2.

Individual predictions submitted for the SAMPL3 host–guest blind prediction challenge. The numbers indicated in the top left corner of each plot corresponds to the submission ID given in Table 2. Host1 systems are indicated by black open circles, whereas hosts 2 and 3 are indicated by red open squares. The blue dotted lines indicate an error range of ±2 kcal/mol
Overall, the predicted binding affinities suffer from high root-mean-square errors (RMSE), low correlations, or both. The RMSEs vary from 1.4 to 45.2 kcal/mol, and the correlation coefficients (R 2) vary from 0.01 to 0.94. The correlation coefficients tend to be higher for the host H2 and host H3 cases than for host H1, but the significance of this pattern is not strong, because the number of data points for H2 and H3 systems is only four, compared to seven data points for H1 system. Moreover, since only a subset of methods was applied to the hosts H2 and H3, it is not clear whether the higher correlations are indicative of the efficacy of these particular methods or of the relatively tractability of the systems. More generally, it is difficult to determine the basis for the differences among the various results with any confidence. One reason is that the submissions varied non-systematically in their choice of force-field parameters and atomic partial charges, solvation models, and conformational sampling methods.
The solvated interaction energy (SIE) model has the lowest RMSE, with better correlation (R 2 = 0.77) for the hosts H2 and H3 than for the host H1 (R 2 = 0.44). Interestingly, this method yielded similar affinities when host H1 was treated as uncharged (submission 5). Interestingly, other methods that used the same GAFF/AM1-BCC force field parameters as the SIE calculations (submissions 6, 7, 13 and 14) were not as accurate. More generally, we could not identify a clear advantage for any particular charge set, force-field, solvation model, or free-energy calculation method. Indeed, seemingly similar methods like TI, BAR, and FEP, combined with similar parameters, produced quite different results, suggesting unexpectedly that the details of the free-energy method might have contributed to the differences among these predictions. For example, submissions 13 and 14 used identical energy models, but their respective RMSEs were 4.0 and 11.3 kcal/mol and their correlation coefficients were 0.34 and 0.81. In principle, the differences in computed free energies are attributable to some combination of differences in energy model and differences in conformations sampled. However, in the absence of information about the predominant conformations found by the various methods, or of a detailed breakdown of the binding free energies into terms like the changes in internal energy, solvation energy, and entropy on binding, it seems impossible to delineate the chief sources of variation among the different predictions. It is also unclear whether or how the correction for standard concentration [59], which normally yields a constant offset, was accounted for in some submissions.
Host1 predictions
The protonation states of the host H1 systems under the experimental conditions are uncertain and might change on binding. In particular, the mutual proximity of the four carboxyl groups on the host may reduce their acidity, allowing some of them to be protonated; and several of the guests contain anilino groups of uncertain pKa. Most of the calculations are based on the standard pKas of the chemical groups involved and assume no change in protonation on binding.
Setting aside the calculations for a moment, one may observe that the experimental affinity trends of the host H1 cases are intuitively reasonable. Thus, the host, whose charge may be as great as −4, binds the neutral guest G4 with lowest binding affinity (−4.17 kcal/mol), the +1 guests (G1, G2, G3, G5, and G7) with intermediate affinity (−6 to −8 kcal/mol), and the +2 charge guest G6 (at pH 7.0 based on standard pKa’s) with highest binding affinity (−10.7 kcal/mol). The sizes of the guests correlate with their charges, so the weakest affinity also corresponds to the smallest guest and the greatest affinity to the largest guest. A majority of the submissions capture this general trend, but they also tend to invert the affinity rankings within the group of +1 guests (G1, G2, G3, G5, and G7), resulting in poor overall correlation coefficients. Two submissions (7 and 14), captured the relative affinities of +1 charged guests somewhat better, resulting in an overall correlation coefficient of 0.8. However, these methods had RSMEs greater than 10 kcal/mol. The large mean error of submission 14 results primarily from a large error for guest G6 (+2); the authors retrospectively attribute this to a setup error (W. Yang, personal communication). It is not clear why these methods ranked the +1 guests relatively well, while others using similar force-field parameters showed anti-correlation for the +1 guests. The fact that host H1 is flexible enough to undergo significant conformational changes might play a role, as different methods might have sampled different conformers.
Host2/host3 predictions
The protonation states of hosts H2 and H3 and their guests are relatively unambiguous, and the molecules are also rather rigid. Both hosts were studied with the same set of guests. The key trend observed in the experimental data is that the guests bind more tightly to the larger host (host H3) than to the smaller one (host H2), and a majority of the submissions captured this trend. Additionally, guest G8 binds host H2 more tightly than guest G9 does, while the converse is true in the case of host H3. Only 4 of the 12 submissions captured this pattern, but the differences in measured binding free energies were less than 1.5 kcal/mol, which is likely well within the range of uncertainty of the energies of current force fields.
More broadly, the RMSEs of the computed binding affinities submitted range from 1.4 and 45.2 kcal/mol, with correlation coefficients between 0.14 and 0.94. Encouragingly, 9 of the 14 submissions show correlations greater than 0.7, despite large RMSEs. Although the SIE model has the lowest RMSE, a method based on a quantum mechanical (QM) energy model has the highest correlation coefficient, 0.94, and clearly captures the key trends in the data. Interestingly, when changes in entropy are dropped from this method, the correlation coefficient drops to 0.64. Also, calculations using the same basic approach as the quantum method, but now using MMFF with AM1-BCC charges (submissions 15 and 16), show similarly high correlation coefficients, greater than 0.9. Much as observed in the host H1 submissions, some binding affinities computed with the same force field and similar continuum solvent models as those of the relatively successful SIE approach incurred significantly larger RMSEs and lower correlation coefficients, and indeed anti-correlated with the measured affinities.
Discussion
This paper summarizes the results of the first blind prediction challenge for host–guest binding affinities. The availability today of host–guest systems with binding affinities similar to those of protein–ligand systems provides an important opportunity to test and ultimately improve physics-based models of binding, in a setting where computational demands are less limiting than they are in the case of proteins. In particular, the challenge of conformational sampling is dramatically reduced for relatively rigid, low molecular weight hosts. New host–guest affinities were measured to enable the present study, and a number of research groups had the courage to submit blind predictions. The participants used a wide variety of approaches ranging from simple docking to extensive molecular dynamics based free-energy calculations.
Sources of accuracy and error
Given the relative simplicity of the host–guest molecular systems, it is somewhat surprising that none of the predictions agree particularly well with the experimental data. In addition, the various methods provide a rather wide spread of results. One fundamental question is whether or not the various methods sampled essentially the same set of conformations. If they did—and one may hope this is the case, given the small sizes of these systems—then the differences among the methods should be attributable primarily to differences in the energy models they used. On the other hand, models with similar energy functions were observed to yield significantly different results (e.g., methods 13 and 14), suggesting significant differences in conformational sampling. It is also striking that the SIE approach [33] provided some of the most accurate results, despite the fact that conformational sampling in this method is limited to extensive sampling of the ligand, while the receptor is kept rigid except for a final energy-minimization step. There was no clear advantage to using more elaborate sampling methods, such as molecular dynamics simulations or the Tork conformational search method [58]. One possible explanation of this broad observation is that conformational sampling might amplify the effects of errors in the energy models by putting too much statistical weight on unrealistic conformations. It is also worth noting that only the SIE method applies empirical weighting coefficients to its computed energy terms. It is intriguing that these coefficients appear to work reasonably well, despite having been fitted based on protein-small molecule data [33].
During the workshop discussion, the protonation states of the host H1 systems emerged as a key source of uncertainty in the calculations. Although the participants were aware of this issue, almost all predictions were based on the simplifying assumption that the protonation states are identical in the bound and unbound conformations, and the charge states were set based on the standard pKa of the different chemical groups in solution. In a few cases where the binding free energy computed for the anilino guests was greater than zero, the participants considered alternate protonation states. There are actually two issues here. In addition to uncertainty in the baseline pKas of the various ionizable groups, especially the four host carboxyls, it is also possible that protonation states change significantly on binding. Indeed, there is substantial experimental evidence for pKa shifts induced by cucurbituril binding [17]. In general, it is observed that cucurbiturils favor binding of the protonated form of the guest, and therefore induce increases in pKa (pK bounda − pK unbounda ) ranging up to +4.5 units; negative shifts as great as −1.5 units have also been observed [17]. The positive pKa shifts of bound guest molecules are for standard, electrically neutral cucurbiturils and, presumably will only be increased for host H1, with its four carboxyls. Of particular relevance here is the pKa shift of the aniline group of 2-aminoanthracene from 4.0 to 7.1 upon binding to cucurbit[7]uril [60]. Also, guests G2 (procaine) and G7 (tetracaine) exhibit 1.22 and 1.91 units of pKa shift, respectively, upon binding to cucurbit[7]uril [61]. Three out of seven guests for the host H1 system contained an aniline group, and therefore the choice of protonation state based on standard pKa could have significantly affected the predicted binding affinities. It is important to note that, to the extent that uncertainties in protonation states are sources of error in the present calculations, the present comparisons are not informative regarding the quality of the force fields used and the adequacy of conformational sampling. Thus, for future rounds of host–guest blind predictions, it may be helpful to steer clear of systems posing this issue, or to be sure of including some without it. Since protonation state issues were not prominent for the host H2 and H3 cases, it is unfortunate that a number of groups in this initial round were unaware of these systems until it was too late to process them before the workshop. Although the predicted binding affinities for hosts H2 and H3 still deviated significantly from the experimental values, the correlation between experiment and computation was, arguably, better for these systems.
The possibility that protonation states and shifts contributed to the computational errors observed here is not necessarily surprising, but it is somewhat sobering, given that similar issues frequently arise in protein–ligand binding because of the high density of charged amino acids in the catalytic sites of enzymes and other protein binding sites [62, 63]. Enzyme active sites in particular frequently contain catalytic histidine residues whose pKas are poised near physiological pH, making their charge states both uncertain and changeable [63]. Although the theoretical framework for coupled protonation and binding equilibria has long been known [64, 65], it is not clear whether current computational methods for modeling protonation equilibria are sufficiently reliable to enable accurate affinity predictions. Prediction of pKas based on physical models has a long history and we recommend several excellent reviews on this topic [66–68]. The pKa Cooperative [21], a blind challenge for structure-based computational methods for predicting pKa values and electrostatic energies in proteins, is notable in this context.
The heart of these binding affinity calculations lies in the energy model itself, i.e., the force field parameters and the solvation model. All participants in the present study used generalized force-field parameters, such as GAFF [38] with AM1/BCC charges [45, 46], except for one group which used a quantum mechanical Hamiltonian. Interestingly, the quantum mechanical results correlated relatively well with experiment, despite overestimating the affinities considerably. Currently, a large amount of benchmarking data is being made available by computational chemists using high-level quantum mechanical models [69–74], and these are likely to be helpful in improving the current generalized force-fields. For instance, Goodman et al. [75] have recently compared several classical generalized force-fields against some of the above mentioned quantum–mechanical benchmarking data and shown that the mean errors in interaction energies are 2 kcal/mol or more, even for seemingly simple, fragment-like molecules. Current protein force-fields (OPLS [37] and AMBER [76]) perform significantly better against similar benchmarks, with mean errors in interaction energies less than 1 kcal/mol [72]. It would appear that, although force-fields for proteins are already well optimized, there may be significant room for improvement of the generalized force fields used for the broader chemistries of drug-like ligands and host–guest systems. Indeed, it may well be that force field errors underlie many of the problems observed in the present exercise.
Changes in solvation free energy also contribute significantly to the computed host–guest binding affinities [6, 10]. Interestingly, although explicit solvent models may be regarded as the gold standard, predictions here using an explicit solvent model did not outperform those that relied on faster continuum methods. In fact, the best-scoring SIE model used the Poisson-Boltzmann surface area (PBSA) implicit solvent model [51]. The existence of other differences among the models makes it impossible to draw definite conclusions about the relative merits of the different solvent models. These broad observations suggest that implicit solvation models will continue to play an important role in models for binding.
Finally, it is of interest to consider whether different computational definitions of the bound state of the various complexes could lead to significant differences among the various calculations and potentially to deviations from experiment. The appropriate definition of the bound state will be unambiguous in systems with a single, deep well in the host–guest potential of mean force [3, 59, 77]. However, the situation can in principle become more complicated, such as if the energy well is shallow or if there are multiple energy wells of similar depths, especially if some of these have the guest positioned outside the cavity of the host.
Implications for protein–ligand modeling
It is interesting to consider how the results of this community exercise bear on the calculation of protein-small molecule binding affinities, particularly in connection with computer-aided drug design. The underlying statistical mechanical principles of binding are expected to be the same for host–guest and protein–ligand systems, and the same physical forces including electrostatic and van der Waals interactions, hydrogen bonding, and solvation, are at play in both settings. However, as noted above, calculations for proteins, rather than hosts, benefit from the availability of more refined force field parameters. On the other hand, it is still necessary to assign force field parameters to small, drug-like molecules, and this could be an important source of error in protein–ligand modeling. Models of both protein–ligand and host–guest systems also encounter the challenge of dealing with protonation states, and issues of solvation models appear similar in both settings. Perhaps the chief difference, then, is that proteins have far more degrees of freedom, so achieving adequate conformational sampling is expected to be far more challenging. It is perhaps relevant that one message of the present study is that more conformational sampling does not necessarily lead to more accuracy. Nonetheless, it is unlikely on physical grounds that calculations without significant conformational sampling can ever be highly accurate. Overall, given the similarities between protein–ligand and host–guest binding, it is hard to imagine that reliable results can be achieved for proteins without also achieving them for host–guest model systems. Because host–guest systems have the added merit of simplicity, they represent informative and tractable test systems for future methods development.
Directions
As evidenced by the special papers in this issue, the results of this blind prediction challenge are leading to re-evaluations of the various methods that will hopefully lead to better results in the future rounds of SAMPL, not only for the host–guest systems but also for protein–ligand binding. The SAMPL3 host–guest binding affinity challenge was the first step in a collaborative process of confronting experiment with computation, and the blind prediction setting is an excellent way to mimic the challenges faced by today’s pharmaceutical industry.
The main goal of this blind prediction challenge was to identify the key problem areas that need further attention. The range of computational methods used here by the participants was informative in assessing the strengths and weaknesses of each of the methods. However, due to the varied modeling choices made by the participants, it was impossible to draw definite conclusions regarding the contribution of the different energy components, solvation, force fields, protonation, and conformational sampling, to the errors in predictions.
Future rounds of host–guest blind predictions would be more informative if participants supplied not only their free energy predictions, but also the coordinates of conformations that were sampled, and, where possible, the energy components underlying their computed free energies. A more detailed analysis of these additional data could shed light on the right direction for future improvements. In addition, one of the major sources of confusion was the choice of ionization states, as the different choices made by some participants masked the effects of other methodological differences, such as the force field and sampling method. It might help to understand this problem in more detail if more participants submitted predictions based on different ionization states. Finally, it was unexpected that seemingly similar approaches using identical energy models resulted in very different predictions. In the future rounds of SAMPL blind prediction challenge, it may be useful for participants to make multiple submissions with controlled variations in the choices, as this would also help understand how different methods performed in the hands of different participants.
Overall, progress on any single front, such as force field parameters or conformational sampling, will likely be insufficient for a marked improvement in the accuracy of predictions. Rather, a concerted effort towards all the challenges discussed here will be required. It will also be important to take advantage of other community prediction exercises, such as the community structure–activity resource (CSAR) [78, 79]. This effort focuses on protein ligand systems, and in addition to affinity scoring, the CSAR challenges the participants to accurately predict binding poses, which are compared against high-quality crystallographic structures. Another blind challenge that deserves mention here is the pKa Cooperative [21], which is focused towards advancing the development of structure-based computational methods for computing pKa values and electrostatic energies in proteins. The continued involvement of researchers across the country and possibly the globe in such prospective prediction challenges will speed progress in molecular modeling and lead to improved tools for computer-aided drug design.
References
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–949
Gilson MK, Zhou HX (2007) Calculation of protein-ligand binding affinities. Annu Rev Bioph Biom 36:21–42
Gallicchio E, Levy RM (2011) Recent theoretical and computational advances for modeling protein-ligand binding affinities. Adv Protein Chem Str 85:27–80
Houk KN, Leach AG, Kim SP, Zhang X (2003) Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes. Angew Chem Int Ed Engl 42:4872–4897
Chen W, Chang CE, Gilson MK (2004) Calculation of cyclodextrin binding affinities: energy, entropy, and implications for drug design. Biophys J 87:3035–3049
Chang CE, Gilson MK (2004) Free energy, entropy, and induced fit in host-guest recognition: calculations with the second-generation mining minima algorithm. J Am Chem Soc 126:13156–13164
Liu SM, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L (2005) The cucurbit[n]uril family: prime components for self-sorting systems. J Am Chem Soc 127:15959–15967
Mock WL (1995) Cucurbituril. Top Curr Chem 175:1–24
Mock WL, Shih NY (1983) Host guest binding-capacity of cucurbituril. J Org Chem 48:3618–3619
Moghaddam S, Inoue Y, Gilson MK (2009) Host-guest complexes with protein-ligand-like affinities: computational analysis and design. J Am Chem Soc 131:4012–4021
Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, Inoue Y, Gilson MK (2011) New ultrahigh affinity host-guest complexes of cucurbit[7]uril with bicyclo[2.2.2]octane and adamantane guests: thermodynamic analysis and evaluation of M2 affinity calculations. J Am Chem Soc 133:3570–3581
Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L et al (2007) A synthetic host-guest system achieves avidin-biotin affinity by overcoming enthalpy-entropy compensation. Proc Natl Acad Sci USA 104:20737–20742
Lee JW, Samal S, Selvapalam N, Kim HJ, Kim K (2003) Cucurbituril homologues and derivatives: new opportunities in supramolecular chemistry. Acc Chem Res 36:621–630
Kim J, Jung IS, Kim SY, Lee E, Kang JK, Sakamoto S, Yamaguchi K, Kim K (2000) New cucurbituril homologues: syntheses, isolation, characterization, and X-ray crystal structures of cucurbit[n]uril (n = 5, 7, and 8). J Am Chem Soc 122:540–541
Ma D, Zavalij PY, Isaacs L (2010) Acyclic cucurbit[n]uril congeners are high affinity hosts. J Org Chem 75:4786–4795
Lagona J, Fettinger JC, Isaacs L (2003) Cucurbit[n]uril analogues. Org Lett 5:3745–3747
Macartney DH (2011) Encapsulation of drug molecules by cucurbiturils: Effects on their chemical properties in aqueous solution. Isr J Chem 51:600–615
Kim K, Selvapalam N, Ko YH, Park KM, Kim D, Kim J (2007) Functionalized cucurbiturils and their applications. Chem Soc Rev 36:267–279
Koner AL, Nau WM (2007) Cucurbituril encapsulation of fluorescent dyes. Supramol Chem 19:55–66
Moult J, Fidelis K, Kryshtafovych A, Tramontano A (2011) Critical assessment of methods of protein structure prediction (CASP)-round IX. Proteins 79(Suppl 10):1–5
Nielsen JE, Gunner MR, Garcia-Moreno EB (2011) The pK(a) cooperative: a collaborative effort to advance structure-based calculations of pK(a) values and electrostatic effects in proteins. Proteins 79:3249–3259
Geballe MT, Skillman AG, Nicholls A, Guthrie JP, Taylor PJ (2010) The SAMPL2 blind prediction challenge: introduction and overview. J Comput Aided Mol Des 24:259–279
Allen FH (2002) The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr B 58:380–388
Biedermann F, Rauwald U, Cziferszky M, Williams KA, Gann LD, Guo BY, Urbach AR, Bielawski CW, Scherman OA (2010) Benzobis(imidazolium)-cucurbit[8]uril complexes for binding and sensing aromatic compounds in aqueous solution. Chemistry 16:13716–13722
Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L (2005) The cucurbit[n]uril family. Angew Chem Int Ed Engl 44:4844–4870
Kirkwood JG (1935) Statistical mechanics of fluid mixtures. J Chem Phys 3:300–313
Straatsma TP, Mccammon JA (1991) Multiconfiguration thermodynamic integration. J Chem Phys 95:1175–1188
Bennett CH (1976) Efficient estimation of free-energy differences from Monte-Carlo data. J Comput Phys 22:245–268
Wang F, Landau DP (2001) Efficient, multiple-range random walk algorithm to calculate the density of states. Phys Rev Lett 86:2050–2053
Gallicchio E, Lapelosa M, Levy RM (2010) The binding energy distribution analysis method (BEDAM) for the estimation of protein-ligand binding affinities. J Chem Theory Comput 6:2961–2977
Zheng L, Chen M, Yang W (2008) Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc Natl Acad Sci USA 105:20227–20232
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J Am Chem Soc 120:9401–9409
Naim M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, Drabik P, Sulea T, Bayly CI, Jakalian A et al (2007) Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model 47:122–133
Chang CE, Gilson MK (2004) Free energy, entropy, and induced fit in host-guest recognition: Calculations with the second-generation mining minima algorithm. J Am Chem Soc 126:13156–13164
Head MS, Given JA, Gilson MK (1997) ‘‘Mining minima’’: direct computation of conformational free energy. J Phys Chem A 101:1609–1618
Jorgensen WL, Maxwell DS, TiradoRives J (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 118:11225–11236
Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 105:6474–6487
Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25:1157–1174
Halgren TA (1996) Merck molecular force field.1. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519
Momany FA, Rone R (1992) Validation of the general-purpose Quanta(R)3.2/Charmm(R) force-field. J Comput Chem 13:888–900
Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I et al (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690
Gilson MK, Gilson HSR, Potter MJ (2003) Fast assignment of accurate partial atomic charges: an electronegativity equalization method that accounts for alternate resonance forms. J Chem Inf Comp Sci 43:1982–1997
Banks JL, Beard HS, Cao YX, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR et al (2005) Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem 26:1752–1780
Bayly CI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the resp model. J Phys Chem Us 97:10269–10280
Jakalian A, Bush BL, Jack DB, Bayly CI (2000) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem 21:132–146
Jakalian A, Jack DB, Bayly CI (2002) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem 23:1623–1641
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev B 136, B864
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:1133
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys Rev A 38:3098–3100
Lee CT, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys Rev B 37:785–789
Sitkoff D, Sharp KA, Honig B (1994) Accurate calculation of hydration free-energies using macroscopic solvent models. J Phys Chem Us 98:1978–1988
Qiu D, Shenkin PS, Hollinger FP, Still WC (1997) The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J Phys Chem A 101:3005–3014
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Corbeil CR, Sulea T, Purisima EO (2010) Rapid prediction of solvation free energy. 2. The first-shell hydration (FiSH) continuum model. J Chem Theory Comput 6:1622–1637
Gallicchio E, Paris K, Levy RM (2009) The AGBNP2 implicit solvation model. J Chem Theory Comput 5:2544–2564
Gallicchio E, Levy RM (2004) AGBNP: an analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J Comput Chem 25:479–499
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Chang CE, Gilson MK (2003) Tork: conformational analysis method for molecules and complexes. J Comput Chem 24:1987–1998
Gilson MK, Given JA, Bush BL, McCammon JA (1997) The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J 72:1047–1069
Wang RB, Yuan L, Macartney DH (2005) A green to blue fluorescence switch of protonated 2-aminoanthracene upon inclusion in cucurbit[7]uril. Chem Commun 5867–5869
Wyman IW, Macartney DH (2010) Host-guest complexations of local anaesthetics by cucurbit[7]uril in aqueous solution. Org Biomol Chem 8:247–252
Bate P, Warwicker J (2004) Enzyme/non-enzyme discrimination and prediction of enzyme active site location using charge-based methods. J Mol Biol 340:263–276
Bartlett GJ, Porter CT, Borkakoti N, Thornton JM (2002) Analysis of catalytic residues in enzyme active sites. J Mol Biol 324:105–121
Wyman J (1965) Binding potential a neglected linkage concept. J Mol Biol 11:631
Schellman JA (1975) Macromolecular binding. Biopolymers 14:999–1018
Bashford D (2004) Macroscopic electrostatic models for protonation states in proteins. Front Biosci J Virtual Library 9:1082–1099
Gunner MR, Mao J, Song Y, Kim J (2006) Factors influencing the energetics of electron and proton transfers in proteins. What can be learned from calculations. Biochim Biophys Acta 1757:942–968
Garcia-Moreno EB, Fitch CA (2004) Structural interpretation of pH and salt-dependent processes in proteins with computational methods. Methods Enzymol 380:20–51
Goerigk L, Grimme S (2010) A general database for main group thermochemistry, kinetics, and noncovalent interactions—assessment of common and reparameterized (meta-)GGA density functionals. J Chem Theory Comput 6:107–126
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41:157–167
Rezac J, Riley KE, Hobza P (2011) S66: a well-balanced database of benchmark interaction energies relevant to biomolecular structures. J Chem Theory Comput 7:2427–2438
Berka K, Laskowski R, Riley KE, Hobza P, Vondrasek J (2009) Representative amino acid side chain interactions in proteins. A comparison of highly accurate correlated ab Initio quantum chemical and empirical potential procedures. J Chem Theory Comput 5:982–992
Jurecka P, Sponer J, Cerny J, Hobza P (2006) Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys Chem Chem Phys 8:1985–1993
Goerigk L, Grimme S (2011) Efficient and accurate Double-Hybrid-Meta-GGA density functionals-evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent Interactions. J Chem Theory Comput 7:291–309
Paton RS, Goodman JM (2009) Hydrogen bonding and pi-stacking: how reliable are force fields? A critical evaluation of force field descriptions of nonbonded interactions. J Chem Inf Model 49:944–955
Duan Y, Wu C, Chowdhury S, Lee MC, Xiong GM, Zhang W, Yang R, Cieplak P, Luo R, Lee T et al (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24:1999–2012
Mihailescu M, Gilson MK (2004) On the theory of noncovalent binding. Biophys J 87:23–36
Dunbar JB, Smith RD, Yang CY, Ung PMU, Lexa KW, Khazanov NA, Stuckey JA, Wang SM, Carlson HA (2011) CSAR benchmark exercise of 2010: selection of the protein-ligand complexes. J Chem Inf Model 51:2036–2046
Smith RD, Dunbar JB, Ung PMU, Esposito EX, Yang CY, Wang SM, Carlson HA (2011) CSAR benchmark exercise of 2010: combined evaluation across all submitted scoring functions. J Chem Inf Model 51:2115–2131
Acknowledgments
This work was supported in part by Grants GM61300 from the NIGMS to M.K.G, CHE-1110911 from the NSF to L.I., Robert A. Welch Foundation (F-1621) to C.W.B, and NSF grant CHE-0748483 to A.R.U. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the National Institutes of Health. We thank Drs. Emilio Gallicchio, Enrico Purisima and Wei Yang for corrections and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Muddana, H.S., Daniel Varnado, C., Bielawski, C.W. et al. Blind prediction of host–guest binding affinities: a new SAMPL3 challenge. J Comput Aided Mol Des 26, 475–487 (2012). https://doi.org/10.1007/s10822-012-9554-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-012-9554-1