
Abstract
Within the framework of the 6th physical property blind challenge (SAMPL6) the authors have participated in predicting the octanol–water partition coefficients (logP) for several small drug like molecules. Those logP values where experimentally known by the organizers but only revealed after the submissions of the predictions. Two different sets of predictions were submitted by the authors, both based on the COSMOtherm implementation of COSMO-RS theory. COSMOtherm predictions using the FINE parametrization level (hmz0n) obtained the highest accuracy among all submissions as measured by the root mean squared error. COSMOquick predictions using a fast algorithm to estimate σ-profiles and an a posterio machine learning correction on top of the COSMOtherm results (3vqbi) scored 3rd out of 91 submissions. Both results underline the high quality of COSMO-RS derived molecular free energies in solution.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The octanol–water partition coefficient (its common logarithm often abbreviated as logKow, logPow or just logP) of a molecule is a physico-chemical property of particular relevance. It serves as a general descriptor for the hydrophilic versus the lipophilic character of a compound and is used for example in the context of the estimation of a drug’s distribution between hydrophilic and lipophilic compartments within the human body. Apart from pharmacology, its usefulness extends also to many other areas such as environmental science, agrochemistry and toxicology. Since the pioneering work of Hansch in the field of partition coefficients and quantitative structure property relations (QSPR) [1], numerous methods have been developed to predict those quantities, based on different model assumptions, with different accuracies but also differing domains of applicability regarding the involved molecular functional groups [2]. This points to a more general problem: the ambiguity in the choice of evaluation data poses a serious issue for the fair assessment of any physico-chemical property method. Therefore, blind prediction challenges with experimental data unavailable to all participants provide the rare opportunity to benchmark a number of methods against each other and to learn and improve approaches based on the insights of this endeavour. This is addressed by the SAMPL series of blind challenges currently hosted by the Drug Design Data Resource initiative (D3R) [3] and taking place first in 2008 [4].
Among the more recent ones, the SAMPL5 challenge [5] was targeted at the logarithmic distribution coefficient of drug-like compounds between water and alkane, i.e. the partitioning between the essentially most different liquid phases occurring in nature. This partition coefficient is mainly relevant for life science in the context of the estimation of the permeability of molecules through cell membranes, which in their center are essentially pure alkane like. The COSMO-RS method provided the most accurate predictions in that challenge [6].
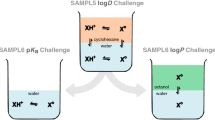
The SAMPL6 challenge considers the somehow related logarithmic octanol–water partition coefficient. In contrast to pure alkane, octanol does provide some polarity and is more representative of typical fatty compartments in human bodies and animals. The challenge was separated into two parts to better reflect the two main aspects of the distribution of a molecule between water and octanol. The first part of the competition finished in early 2018 and dealt with dissociation of the compounds in the aqueous phase and hence the prediction of pKa-values [7, 8]. Please note, that the pKa predictions having the lowest RMSE were based on COSMO-RS computed free energies of solvation [9]. The second part of the SAMPL6 challenge, which is the topic of this work, was solely focused on the prediction of logP values, i.e. the partition of the unionized species between water and octanol.
Experimentally, neutral-compound logarithmic partition coefficients (logP) were collected by the organizers using potentiometric logP (pH-metric) measurements [10,11,12]. Technically, the measured pKa values from the first part of the challenge enter the evaluation of the experiment in order to correct for the effects of acidic or basic groups. Due to experimental issues e.g. related to too low solubilities only 11 out of the original 20 drug-like molecules could be measured and remained for prediction (Fig. 1).

Molecular structures of the 11 protein kinase inhibitor fragments used for the logP prediction challenge
Finally, two sets of logP predictions were submitted by the authors. Among a total number of 91 submissions, COSMOtherm using the FINE parameterization scored the lowest root mean squared error (RMSE) and mean absolute deviation (MAE). COSMOquick based predictions using an empirical correction term based on machine learning (ML) on top of lower level COSMOtherm calculations (estimated σ-profiles at the TZVP level) came in third according to the competition metric.
COSMO-RS theory
The Conductor like Screening Model for Realistic Solvation (COSMO-RS) was developed by Klamt in 1995 basically to overcome the deficiencies of classical implicit solvation models [13]. Intermolecular interactions in COSMO-RS are defined via surface segments derived from the screening charge on the surface of an isolated molecule placed within a virtual conductor. An implicit solvation COSMO [14] calculation using density functional is carried out in order to obtain the σ-surface and the resulting data is finally saved in a COSMO file for further use. Technically, those COSMO files can be stored in a database that allows to use pre-computed σ-surfaces for common molecules such as drugs, solvents and polymers.
Instead of an ensemble of interacting molecules as considered in a classical molecular mechanics simulation, COSMO-RS describes intermolecular interactions from an ensemble of pair-wise interacting surface segments. An important simplifying assumption being made is that all segment wise interactions in the liquid state are possible, which leads to a dramatic improvement in the overall efficiency of the thermodynamic sampling. COSMO-RS takes into account most relevant intermolecular interactions, such as Van der Waals interactions, hydrogen bonding and also short-ranged electrostatic interactions. Except from the Van der Waals interaction, which are based on element specific contributions, all interactions arise from the pairwise contacts of the surface segments.
The segments of the σ-surface may be projected into a histogram of equally binned charges densities, the σ-profile of a compound.
Just by visual inspection of the σ-profile it is possible to derive some qualitative information about the solvation characteristics. The profiles displayed in Fig. 2 show the difference in the polar, i.e. hydrogen bond donating and accepting regions between the relatively hydrophilic compound SM11 and the more lipophilic SM02. According to its σ-profile, compound SM11 (Fig. 2, dashed line) shows significantly more polar surface area within the regions having σ < − 0.01 e/Å, σ > 0.01 e/Å, respectively, whereas SM02 (solid line) presents a stronger peak at the hydrophobic region (σ ~ 0.0 e/Å).

The σ-profiles of the two SAMPL6 compounds having the lowest (SM11) and the highest (SM02) predicted octanol–water partition coefficient. Only the profiles of the lowest energy conformers are shown
Finally, quantitative chemical potentials are derived from rigorous statistical thermodynamics using the surface segment based molecular interactions finally providing access to all kind of different physico-chemical properties in solution. For more details of the approach and the definitions of the energetic interactions we refer to a concise review by Klamt [15].
Computational methods
The logP dataset of the challenge consists of 11 small to medium sized protein kinase inhibitor fragments which combine several functional groups having at least one basic or acidic site. The logP data extends about 2 log units from roughly 2 (SM14) to 4 (SM02).
Due to the fact, that pure logP values of the neutral species were extracted from the experiment, no dissociation or association effects related to acidic or basic sites had to be taken into account during the modelling.
All density functional theory calculations were carried out with the BP86 functional [16, 17], using a TZVP [18] basis set for geometry optimisation (BP-TZVP-COSMO), and where specified, a def2-TZVPD basis set [19] single point calculation on top of the TZVP optimized geometry (FINE-COSMO), always using the COSMO solvation [14] scheme. A systematic tautomer search for the eleven compounds was carried out using the COSMOquick software [20] by systematic (de)protonation of the σ-surface hotspots at the semi-empirical AM1/COSMO level [21] and subsequent BP-TZVP-COSMO based COSMOtherm calculations. However, no significantly populated tautomeric forms were detected for the compounds under consideration, neither in water nor in octanol. Two compounds, SM09 and SM12, which originally were defined as salts, were modelled as neutral non-salt molecules.
For all compounds independent sets of relevant conformations were computed with the COSMOconf 4.3 workflow [22]. The quantum chemistry calculations of COSMO surfaces were done on the FINE-COSMO level based upon BP-TZVP-COSMO optimized geometries to match the parameterization used in COSMOtherm. The quantum chemical calculations were done with the TURBOMOLE 7.3 quantum chemistry software [23].
The distribution coefficient logP between the pure solvent phases water and octanol was computed for all solutes using the COSMO-RS method as implemented in COSMOtherm, according to the following general procedure (in the context of the challenge this submission is referenced as hmz0n).
For all compounds, relevant conformations in the liquid phase were used as previously generated by COSMOconf. Partition coefficients logP were calculated from the differences of the infinite dilution chemical potentials μ of the solute × in the solvents octanol (phase 1) and water (phase 2):
Molar volumes V were used to convert from the mole fraction basis to a molar concentration-based framework. For all compounds various conformational states were taken into account by calculation of the total chemical potentials from the logarithm of the conformational partition function. The solvent octanol was treated as wet octanol, i.e. assumed to have a water mole fraction content of 27.4% [24]. All COSMOtherm calculations were done with the BP_TZVPD_FINE_19 parameterization [13, 25, 26].
Prior to the release of the experimental measurements, accuracies of the COSMOtherm predictions were estimated according to the following assumptions: the accuracy of the COSMOtherm BP_TZVPD_FINE_19 parameterization for the prediction of logarithmic partition coefficients is 0.35 log units (RMSD). For 262 non-polar solvent/water partition coefficients appearing in our parameterization and validation sets with exp. data taken from the Biobyte database [27] the RMSD is 0.4 log units. Taking into account that the compounds in these datasets are in average smaller by a factor 2 than the compounds considered in the SAMPL6 challenge, and assuming uncorrelated errors, the estimated accuracy of the prediction of logP octanol–water is in the order of 0.5 log units. In retrospective, this estimation seems to be slightly too conservative with regard to the final accuracy of the predictions with an RMSE of 0.38 logP units which coincides well with the original 0.35 log units accuracy of the parameterization.
A second set of predictions based on an alternative approach omitting explicit quantum chemical calculations was submitted (reference 3vqbi). For all compounds approximated σ-surfaces were generated directly from the given SMILES strings using a database of about 200.000 pre-computed molecules at the BP-TZVP-COSMO level with the COSMOquick 1.7 software and the COSMOfrag algorithm [20, 28]. Hence, no additional costly quantum-chemical calculations was required.
The distribution coefficient logP between the pure solvent phases water and wet 1-octanol was computed for all solutes using the COSMO-RS method as implemented in COSMOtherm, in a similar fashion as for submission hmz0n.
Partition coefficients logP for all solutes were calculated from the infinite dilution chemical potential differences within the solvents water and octanol. All COSMOtherm calculations were done with the BP_TZVP_18 parameterization [13, 25, 26]. This parameterization is using a somewhat smaller basis set for the DFT energies and COSMO cavity calculations than the higher level BP_TZVPD_FINE_19. As by inspection of the atomic similarities the quality of estimated σ-profile of structure SM08 was rather low, the pure COSMO file at the TZVP level was used instead of the approximation for this compound.
Finally, an empirical correction was added via machine learning using a decision tree ensemble (Stochastic Gradient Boosting [29] via the XGBoost library [30]). The correction term was trained on a curated experimental dataset of about 11,000 logP values taken from the physprop database [31] based on ten different descriptors. As target vector the difference between the predicted COSMOtherm and experimental data points was used. The following set of COSMOquick based descriptors has been selected for the construction of the decision trees, with some focus on the meaningfulness of the descriptors: N_amino (the number of secondary or tertiary aliphatic amino groups in the compound), mu_gas (chemical potential in the gas), M3 (third sigma moment as derived from the sigma-profile), h_hb (hydrogen bond part of the enthalpy), rotatable_bonds (number of rotatable bonds), conjugated_bonds (number of conjugated bonds), Macc4 (4th order hydrogen bond acceptor sigma moment), mu_water (chemical potential in water), internal_hbonds (number of potential internal hydrogen bonds) and alkylatoms (number of carbon atoms belonging to alkylgroups). Parameters have been optimized using an “early stopping” technique during fivefold cross-validation. Early stopping allows to find the optimal number of boosting iterations by simultaneously monitoring a validation set during training/cross-validation. This logP prediction procedure is a slight modification of the one published with the last release (COSMOquick v1.7), which uses a standard Random Forest instead of gradient boosting for model building, however the outcome for the logP challenge is rather similar.
Most important descriptors as shown in Fig. 3 are the number of aliphatic amino groups, the number of conjugated bonds, the chemical potential in water and the number of potential internal hydrogen bonds.

Variable (permutation) importance for the logP correction term as computed by the R randomForest package. The importance of a specific variable is defined as the percentage increase in the mean squared error (incMSE) upon random shuffling this very variable and subsequent property prediction
The final predicted logP values are computed as the simple sum of the COSMOtherm prediction and the decision tree ensemble based correction:
The cross-validation accuracy during training, was about 0.5 log units, whereas the accuracy on a structurally similar test set, as compared to the SAMPL6 structures, which was excluded from training, was significantly lower. As a conservative estimate the accuracy of the prediction was assumed to be of a similar size as for the cross-validation. As compared with the final RMSE of 0.41 this seemed to be a slight overestimation of the error of the approach.
Results and discussion
In order to evaluate in particular the models based on machine learning and to decide which ones finally being used for submission, several test datasets were constructed using experimental data from the literature. The first test set (test set 1) was focused on the experimental setup and used a data set of partition coefficients measured by different methods from Slater et al. [12]. This collection contains about 40 small acid and basic molecule including a few drugs and for most compounds a set of three independent logP values measured by HPLC, by potentiometric methods, and as collected data from the literature. This collection was chosen because it contains logP values measured with a similar approach as for the SAMPL6 challenge and due to its reliability. Nevertheless, two compounds of the set, celiprolol and acebutolol were treated as outliers due to their strong deviation from the COSMOtherm predictions and removed from the set.
Unfortunately, most compounds contained in test set 1 are structurally unrelated to the challenge compounds, hence a second test set was generated using a substructure search on the physprop database based on the core functional groups (mostly quinazolines) of the challenge compounds. After removing five apparent outliers based on the deviation from practically all employed models, a set of 32 compounds remained (test set 2). This structurally related test set revealed some advantage of the high level FINE parameterization predictions versus the TZVP level predictions and hence the decision was made to only submit the former to the challenge.
The results for the test sets 1 and 2 are summarized in Table 1. Given the fact that the final COSMOtherm predictions outperformed COSMOquick on the challenge compounds, it looks like that the COSMOquick results on the test set are somewhat overfitted, even though the test set compounds themselves were of course excluded from fitting procedure. The COSMOtherm predictions somewhat underperform for the test sets. Indeed, the improvement for the specific compounds of this challenge as compared to the test set compounds is somewhat typical for COSMOtherm, which is particular predictive for novel and unseen structures where most of specifically fitted methods such as QSPRs are prone to fail. Another reason for the different performance of COSMOquick for the test set and final predictions is the fragmentation process used to generate the approximate (meta) COSMO files. The structures of the logP challenge are not as well represented by the COSMOquick approach as the test set compounds are. In fact, using the original COSMO files, i.e. the exact σ-profiles, would have resulted in an RMSE = 0.35 for the COSMOquick model. Supplemental Table S1 shows the influence of the σ-profile estimation on the logP predictions, which is much smaller for the test sets than for the SAMPL6 compounds.
After the test set evaluations, two models based on COSMOtherm (hmz0n) and on COSMOquick employing an a posteriori ML correction (3vqbi) were used to create the final predictions for the challenge. The results of the two submissions as compared against the experiment are shown in Fig. 4, the metrics (root mean squared error RMSE and Pearson correlation coefficient R2) are shown in Table 2. In terms of the RMSE both models perform similar with COSMOtherm being slightly ahead. Given the somewhat small sample size and the narrow exptl. logP range of about 2 units, the correlation coefficient is a less meaningful quantity here. Consequently, only the RMSE was used by the organizers to rank the individual submissions. Among the 91 submissions made by a diversity of methods, COSMOtherm and COSMOquick made predictions are ranked at the 1st and 3rd position, respectively. Interestingly, the predictions themselves seem to be somewhat different and less correlated as to be expected, as shown in Fig. 4 and as demonstrated by a squared correlation coefficient of R2 = 0.76.

Predicted versus experimental logP values for COSMOtherm and COSMOquick of the SAMPL6 logP challenge. A corridor of 0.5 logP units is shown representing the conservatively estimated accuracy of the predictions
Individual predictions are shown in Table 3. The strongest mismatch with the experimental data for COSMOtherm is SM13, where the predictions (3.84) is nearly 1 log unit above the experiment (2.92). In fact, most of the top submissions predict a logP value significantly higher for this species, which may rise some speculation concerning the accuracy of the experimental value for SM13 (see also Supplemental Fig. S1). Impurifications as a cause could be ruled out by the organizers based on experimental MS and NMR data [32]. Whether this noticeable deviation is caused on the experimental side, e.g. via aggregation in the aqueous phase or due to the low solubility, or on the modelling side, could not be resolved at the time of preparation of this work.
The largest outlier for the COSMOquick based submission is for SM15 where the experimental logP value is underestimated by about 0.8 log units. This is also the compound with the lowest σ-profile quality, i.e. the lowest overall fingerprint similarity with the COSMOquick database fragments. The prediction using a full COSMO file (logP = 2.67) is significantly closer to the experiment (logP = 3.07).
Conformational effects on logP
One of the main differences between the COSMOtherm and the COSMOquick approach is the complete neglect of conformational effects in the latter. Therefore, it may be interesting to look at the effect of conformations for the logP predictions. For this purpose COSMO files at the BP-TZVP level were generated on top of randomly created structures and compared with results for COSMOconf sampled molecules. Even though the flexibility of the molecules is rather small there is a significant improvement on the overall RMSD for the rigorous conformational sampling. The RMSD based on random sampling (RMSD = 0.45, Table S2) is also larger than for the COSMOquick model (RMSD = 0.41), which possibly learned to take some conformational effects into account implicitly, as can be assumed from the high feature importance of the rotatable bonds and internal hbond descriptor as shown in Fig. 3.
Conclusions
Within the framework of the SAMPL6 logP blind challenge the authors have submitted two COSMO-RS based predictions. The pure COSMOtherm based submission using the FINE parameterization (hmz0n) constitutes the most accurate entry having a root mean squared deviation of 0.39 against the experiment. This prediction was made using the standard software package and no specific adaption, fitting or modification of the COSMOtherm logP module was made for this challenge. Another submission based on an efficient protocol that allows to quickly estimates σ-profiles from a database followed by a machine learned a posteriori correction term (COSMOquick) scored 3rd among 91 total submissions with an RMSE = 0.41 (3vqbi). Interestingly, both submission show a relatively low correlation with each other and hence may be used complementary in doubtful cases. Corroborated by the fact that the chemical potentials used as basis for the logP predictions were not specifically fitted to this compound class, the results of the SAMPL6 demonstrate the high accuracy of COSMO-RS based free energies in solution.
References
Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71:525–616. https://doi.org/10.1021/cr60274a001
Mannhold M, Poda G, Ostermann C, Tetko I (2009) Calculation of molecular lipophilicity: state of the art and comparison of methods on more than 96000 compounds. Chem Cent J 3:O7. https://doi.org/10.1186/1752-153X-3-S1-O7
(2019) Drug design data resource. In: Drug Des. Data Resour. https://drugdesigndata.org. Accessed 1 Feb 2019
Nicholls A, Mobley DL, Guthrie JP et al (2008) Predicting small-molecule solvation free energies: an informal blind test for computational chemistry. J Med Chem 51:769–779
Rustenburg AS, Dancer J, Lin B et al (2016) Measuring experimental cyclohexane-water distribution coefficients for the SAMPL5 challenge. J Comput Aided Mol Des 30:945–958
Klamt A, Eckert F, Reinisch J, Wichmann K (2016) Prediction of cyclohexane-water distribution coefficients with COSMO-RS on the SAMPL5 data set. J Comput Aided Mol Des 30:959–967. https://doi.org/10.1007/s10822-016-9927-y
(2017) SAMPL6—pKa-prediction—overview. In: PKa-Predict.—Overv. https://drugdesigndata.org/about/sampl6/pka-prediction. Accessed 6 Dec 2017
Işık M, Levorse D, Rustenburg AS et al (2018) pKa measurements for the SAMPL6 prediction challenge for a set of kinase inhibitor-like fragments. J Comput Aided Mol Des 32:1117–1138. https://doi.org/10.1007/s10822-018-0168-0
Pracht P, Wilcken R, Udvarhelyi A et al (2018) High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge. J Comput Aided Mol Des 32:1139–1149. https://doi.org/10.1007/s10822-018-0145-7
Avdeef A (1992) pH-Metric log P. Part 1. Difference plots for determining ion-pair octanol-water partition coefficients of multiprotic substances. Quant Struct-Act Relatsh 11:510–517. https://doi.org/10.1002/qsar.2660110408
Avdeef A (1993) pH-Metric log P. II: refinement of partition coefficients and ionization constants of multiprotic substances. J Pharm Sci 82:183–190. https://doi.org/10.1002/jps.2600820214
Slater B, McCormack A, Avdeef A, Comer JEA (1994) PH-Metric logP.4. Comparison of partition coefficients determined by HPLC and potentiometric methods to literature values. J Pharm Sci 83:1280–1283. https://doi.org/10.1002/jps.2600830918
Klamt A (1995) Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J Phys Chem 99:2224–2235. https://doi.org/10.1021/j100007a062
Klamt A, Schüürmann G (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2(1993):799–805. https://doi.org/10.1039/P29930000799
Klamt A (2018) The COSMO and COSMO-RS solvation models: COSMO and COSMO-RS. Wiley Interdiscip Rev Comput Mol Sci 8:e1338. https://doi.org/10.1002/wcms.1338
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100. https://doi.org/10.1103/PhysRevA.38.3098
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824. https://doi.org/10.1103/PhysRevB.33.8822
Schäfer A, Huber C, Ahlrichs R (1994) Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys 100:5829. https://doi.org/10.1063/1.467146
Rappoport D, Furche F (2010) Property-optimized Gaussian basis sets for molecular response calculations. J Chem Phys 133:134105. https://doi.org/10.1063/1.3484283
(2018) COSMOquick 1.7. COSMOlogic GmbH & Co. KG; http://www.cosmologic.de, Leverkusen, Germany
Stewart JJP (1993) MOPAC7. Quantum Chemistry Program Exchange; http://sourceforge.net/projects/mopac7/, University of Texas, Austin, TX, USA
(2018) COSMOconf 4.3. COSMOlogic GmbH & Co. KG; http://www.cosmologic.de, Leverkusen, Germany
(2018) TURBOMOLE V7.3. University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com, Karlsruhe, Germany
Dallos A, Liszi J (1995) (Liquid + liquid) equilibria of (octan-1-ol + water) at temperatures from 288.15 K to 323.15 K. J Chem Thermodyn 27:447–448. https://doi.org/10.1006/jcht.1995.0046
Klamt A, Jonas V, Bürger T, Lohrenz JC (1998) Refinement and parametrization of COSMO-RS. J Phys Chem A 102:5074–5085. https://doi.org/10.1021/jp980017s
(2019) COSMOtherm, Release 19. COSMOlogic GmbH & Co. KG; http://www.cosmologic.de, Leverkusen, Germany
(2007) BioByte Masterfile. BioByte Corporation, Claremont, CA, USA
Hornig M, Klamt A (2005) COSMOfrag: a novel tool for high-throughput ADME property prediction and similarity screening based on quantum chemistry. J Chem Inf Model 45:1169–1177. https://doi.org/10.1021/ci0501948
Friedman JH (2002) Stochastic gradient boosting. Comput Stat Data Anal 38:367–378. https://doi.org/10.1016/S0167-9473(01)00065-2
Chen T, Guestrin C (2016) XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. ACM, San Francisco, California, USA, pp 785–794
EPA (2014) EPI Suite Data. http://esc.syrres.com/interkow/EpiSuiteData_ ISIS_SDF.htm. Accessed 2 Feb 2019
Isik M (2019) Personal Communication
Acknowledgements
The authors acknowledge the organizers for setting up the SAMPL6 challenge and the SAMPL NIH Grant 1R01GM124270-01A1 for the support of the experimental work carried out in this context.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare the following competing financial interest(s): Andreas Klamt, Jens Reinisch and Christoph Loschen are employees of Dassault Systèmes, BIOVIA. Dassault Systèmes commercially distributes software implementations of COSMO-RS (COSMOtherm, COSMOquick) which were used in the present strudy.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10822_2019_259_MOESM1_ESM.zip
Supplementary material 1—All COSMO files generated by Turbomole representing the conformations used by COSMOtherm for the calculation of the logP values. Additional information about the influence of the σ-profile fragmentation process on prediction quality and the role of conformational effects. (ZIP 5158 kb)
Rights and permissions
About this article

Cite this article
Loschen, C., Reinisch, J. & Klamt, A. COSMO-RS based predictions for the SAMPL6 logP challenge. J Comput Aided Mol Des 34, 385–392 (2020). https://doi.org/10.1007/s10822-019-00259-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-019-00259-z