
Abstract
Purpose
In clinical practice, the success of preimplantation genetic testing for monogenic diseases (PGT-M) for thalassemia was hindered by the absence of probands, incomplete family members, or failure in detecting embryonic gene mutation sites. This study aimed to address these issues.
Methods
This retrospective study included 342 couples undergoing PGT-M for α- or β-thalassemia at three reproductive medicine centers from 2019 to 2022. Various methods were used to construct parental haplotypes. A total of 1778 embryos were analyzed and selected for transfer based on chromosomal ploidy and PGT-M results. Follow-up involved amniocentesis results and clinical outcomes.
Results
Haplotypes were established using DNA samples from probands or parents, as well as sibling blood samples, single sperm, and affected embryos, achieving an overall success rate was 99.4% (340/342). For α-thalassemia and β-thalassemia, the concordance between embryo single nucleotide polymorphism (SNP) haplotype analysis results and mutation loci detection results was 93.8% (1011/1078) and 98.2% (538/548), respectively. Multiple annealing and looping-based amplification cycles (MALBAC) showed a higher whole genome amplification success rate than multiple displacement amplification (MDA) (98.8% (1031/1044) vs. 96.2% (703/731), p < 0.001). Amniocentesis confirmed PGT-M outcomes in 100% of cases followed up (99/99).
Conclusion
This study summarizes feasible solutions to various challenging scenarios encountered in PGT-M for thalassemia, providing valuable insights to enhance success rate of PGT-M in clinical practice.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Thalassemia is a common autosomal recessive monogenic disorder worldwide, particularly prevalent in the Mediterranean region, North Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Guangdong and Guangxi provinces in China [1,2,3]. Based on specific defects in the globin genes, thalassemia is primarily classified into two types: α-thalassemia and β-thalassemia. Carrier frequency for β-thalassemia ranges from 1 to 20% in these regions, whereas the carrier frequency of α-thalassemia is comparatively higher, reaching up to 40% in certain Middle Eastern and Indian populations [2].
α-Thalassemia is caused by the deletion or mutation of the α-globin genes, including α1 and α2, located at the end of the short arm of chromosome 16 (16p13.3). Gene deletions account for the majority of α-thalassemia cases, with the Southeast Asian deletion (–SEA) being the most prevalent type [3]. Couples both carrying the (–SEA) genotype face a 25% risk that their offspring will have Bart’s hydrops fetalis syndrome, which can lead to fetal death in utero or at birth [4]. β-Thalassemia results from mutations in the β-globin gene on the short arm of chromosome 11, resulting in reduced (β+) or absent (β0) synthesis of the β-globin chain [5, 6]. The severe anemia and complications associated with β-thalassemia (β+/β0 or β0/β0) include liver damage, heart disease, and endocrine dysfunction [7, 8]. Without regular transfusions, iron chelation, or hematopoietic stem cell transplantation, these patients face a high risk of mortality in early childhood.
Currently, prenatal diagnosis helps prevent severe thalassemia births, but it may lead couples to consider pregnancy termination [8]. Preimplantation genetic testing for monogenic disorders (PGT-M) has been applied for the detection of single-gene diseases prior to embryo implantation, greatly reducing the risk of miscarriage. Next-generation sequencing (NGS) and single nucleotide polymorphism (SNP) arrays are now key methods for PGT-M, enabling both monogenic disease detection and aneuploidy screening [9,10,11]. Using NGS and SNP array–based PGT testing, euploid embryos that are either normal or carriers of target gene can be transferred.
Thalassemia exhibits various pathogenic nucleotide variations, ranging from point mutations to deletions spanning several to tens of kilobases (Kb). Allele dropout (ADO) remains a challenge due to single-cell amplification limitations, posing a risk of misdiagnosis. To ensure accurate embryo diagnostics, it is necessary to construct parental and embryonic haplotypes. However, constructing parental haplotypes can be challenging in certain cases, such as when DNA samples of probands or relatives are unavailable or when the couple’s mutations are de novo. If parental haplotype construction fails, subsequent embryonic SNP linkage analysis cannot be performed. Additionally, when mutation site detection in embryos fails or shows inconsistencies with SNP linkage analysis, there is a lack of clinical data on the accuracy of embryo transfers based on SNP linkage results.
The current clinical experience in PGT-M for thalassemia within the field of assisted reproduction is predominantly confined to single-center studies detailing a limited number of cases [4, 12, 13]. Well-summarized strategies to address a range of complex scenarios have yet to be established. This study is the first large-scale retrospective analysis of real-world PGT-M for α- and β-thalassemia, summarizing the experience in various clinical scenarios. It aims to address the issues in establishing family haplotypes and mutation site testing in PGT-M for thalassemia, providing solutions to enhance the accuracy and success rates in clinical practice.
Methods
Patients
This large-scale, multicenter retrospective study included 342 couples with α- or β-thalassemia who underwent PGT-M at three reproductive medicine centers from 2019 to 2022, with a total of 1778 embryos tested. Each couple provided written informed consent prior to the PGT cycle, and if peripheral blood samples from other family members were required, additional informed consent was obtained. Ethical approval was granted by the relevant committees at each participating center (No. 2024ZSLYEC-323). Due to the retrospective design of the study, there was no need for extra participant declaration consent.
The inclusion criteria were as follows: couples with a high risk of giving birth to children with α- or β-thalassemia, with a clear clinical diagnosis and defined pathogenic gene mutations; both partners met the fertility assessment criteria for PGT-M; intracytoplasmic sperm injection (ICSI) procedures were used; at least one 3BB blastocyst was available; no other conditions precluded in vitro fertilization and embryo transfer (IVF-ET); single blastocyst transfer was guided by preimplantation genetic testing for aneuploidy (PGT-A)/PGT-M results. Exclusion criteria included participation in other studies or any situations deemed unsuitable for this study by the researchers.
Pedigree analysis
In addition to samples from the couple, the samples from neonatal peripheral blood, amniotic fluid/chorionic villi, products of conception tissue, or blood samples from the siblings can be used to establish parental haplotypes. Genomic DNA (gDNA) extracted from the peripheral blood samples serves as a template for panel-based amplification and was performed following enrichment of the target gene, covering a 1Mbp region surrounding the α- and β-thalassemia genes, inclusive of 286 SNP sites upstream and downstream. Next-generation sequencing follows enrichment. The parental haplotypes were constructed by choosing the available SNP loci for linkage analysis. SNPs were deemed informative only if they were heterozygous in the carriers and homozygous in their partners [14].
When DNA samples from probands or relatives were unavailable, parental haplotypes were established through linkage analysis using SNP loci from embryo biopsy samples displaying the mutant haplotype. The process involves directly detecting mutation sites in all embryos. Embryo biopsy samples with the mutant haplotype are identified as reference embryos. Then, by utilizing the SNP sites from the reference embryos, the couple’s haplotypes were constructed (Supplementary Fig. 1).
Simultaneously, direct mutation loci testing of the HBA/HBB genes in the couple’s DNA samples will be performed using gap-polymerase chain reaction (Gap-PCR) or Sanger sequencing to further verify the accuracy of the family pedigree.
Ovarian stimulation and ICSI procedure
Controlled ovarian stimulation and ICSI were performed according to each center’s standard routine procedures. On the 5th and 6th days after oocyte retrieval, the grading of blastocyst quality was assessed according to Gardner’s scoring criteria [15].
Embryo biopsy
Biopsies were performed on blastocysts that had developed to stage 4 on days 5 or 6. The biopsied trophectoderm cells were placed in a collection tube for laboratory testing. Blastocysts were vitrified immediately after the biopsy using a Kitazato vitrification kit (Kitazato BioPharma, Japan).
Embryonic copy number variation analysis
Following the provided instructions, cells from the biopsy were lysed using lysis buffer, and whole genome amplification (WGA) was performed using either the MALBAC (Yikon Genomics, China) or MDA (Qiagen, Germany) methods. The amplified products were used for copy number variant (CNV) detection and PGT-M testing of α- and β-thalassemia. The procedure for CNV detection and analysis was described previously [16]. The CNV library fragments were enriched using the Universal DNA Fragmentation Kit (Yikon Genomics, Shanghai, China), and the library was constructed directly. Sequencing was performed on the Illumina platform (Illumina, USA). The sequence data was processed and analyzed with ChromGo software (Yikon Genomics, China). Similar but not identical reads were required to pass a series of quality assurance metrics. Unique mapped reads were then computed to generate a reference dataset representing relative copy numbers.
For CNV analysis, the bioinformatics quality control standards were a CV value ≤ 0.2 and valid reads > 1 M. Successful amplification refers to the sequencing data passed the quality control standards. Mosaicism ≥ 30%, with a detection limit for segmental aneuploidy of ≥ 4 Mb, was considered aneuploid in CNV analysis.
SNP linkage analysis
Appropriate SNP loci were chosen for couples when parental haplotypes were established successfully, and then SNP linkage analysis was performed on the embryos. Based on the results of the linkage analysis, the embryos were classified as normal, carriers, or affected. For couples where the absence of family samples results in failed haplotype construction, reference embryos were used to help establish family haplotypes by SNP linkage analysis, followed by SNP linkage analysis on the remaining embryos.
Embryonic HBA or HBB gene mutation detection
For embryos from couples with deletional α-thalassemia, Gap-PCR was performed on the amplified DNA products from the embryos, followed by gel electrophoresis. The normal or deletion of the α-globin gene was determined based on the banding pattern observed on the gel [17, 18]. For patients with point mutations in thalassemia genes, polymerase chain reaction (PCR) was used to amplify the target genes in the embryos, and Sanger sequencing was performed on the PCR products. The presence of mutations at specific loci was determined based on the results of Sanger sequencing.
Embryo transfer
Embryos were selected for transfer based on their euploidy status and the absence of high-risk thalassemia. Single embryo was chosen for each transfer. The thawing and transfer procedures followed the standard protocols established by each center.
Clinical outcomes and statistical analysis
The primary outcome was clinical pregnancy, defined as the presence of a gestational sac confirmed by ultrasound examination 28–30 days after the transfer. Amniocentesis was performed during mid-pregnancy.
Continuous variables were expressed as mean ± standard deviation (X ± SD), while categorical variables were represented as ratios or percentages (%). The results of amniocentesis were as the gold standard. Statistical analysis involved calculating amplification success rates and using chi-square tests to compare amplification methods.
Results
This study included 342 couples who requested PGT-M for α- or β-thalassemia at three reproductive centers between 2019 and 2022. In total, 1778 embryos were tested, with the average maternal age being 31.3 ± 4.3 years. Two hundred twenty-six couples were carriers of α-thalassemia, yielding 1197 blastocysts; 112 couples were carriers of β-thalassemia, contributing 568 blastocysts; and 4 couples were carriers of both α- and β-thalassemia, resulting in 13 blastocysts. The median number of embryos obtained per couple was 5, ranging from a minimum of 1 to a maximum of 21 embryos (Table 1).
Haplotype phase
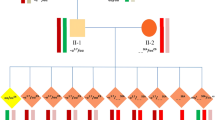
Figure 1 details three scenarios of haplotype analysis, from straightforward (Scenario I) to most challenging (Scenario III). Scenario I is the most straightforward when biological samples from probands were available; Scenario II presents moderate difficulty when the couple lacks probands but provides complete samples from both biological parents; Scenario III poses the greatest challenge, marked by the absence of a proband and a lack of parental DNA samples.

Haplotype analysis from straightforward (Scenario I) to most challenging (Scenario III) in 342 couples. Scenario I: proband DNA samples available; Scenario II: parental DNA samples available; Scenario III: no DNA samples from probands or incomplete parental DNA samples
Scenario I
Among the 342 couples included in this study, 59 (17.3%) fit Scenario I as described above. Samples from the probands included peripheral blood, miscarriage tissue, amniotic fluid, and chorionic villi. The SNP haplotypes linked to the defective chromosomes carried by these couples were successfully confirmed and used to guide subsequent embryo selection (Fig. 1).
Scenario II
Two hundred two couples (59.1%) were categorized under Scenario II. Among these 202 couples, 198 couples (98.0%, 198/202) successfully obtained valid SNP loci for linkage analysis by sequencing both parental samples. However, the SNP haplotypes of four couples could not be determined based on the parental SNP loci. In one couple, the female partner had a de novo mutation (–SEA) in the α-thalassemia gene, whose parents had normal α-globin genes. In the other three couples, referred to as heterozygous families, the genotype of one partner was αα/–SEA, and since both parents were αα/–SEA, it was unclear which disease-causing chromosome was inherited. Among these 4 couples, in one couple with a de novo mutation and two couples with αα/–SEA, the haplotypes were successfully determined through reference embryo SNP loci (Fig. 1; Supplementary Fig. 2). In the remaining couple, by providing samples from two unaffected siblings and one carrier sibling, along with valid SNP loci from the parental samples, the haplotype was successfully determined (Supplementary Fig. 3).
Scenario III
The remaining 81 (23.7%) couples were categorized under Scenario III, wherein probands and either one or both parents’ blood samples were unavailable (Fig. 1). Among them, 40 couples provided peripheral blood samples from siblings, as detailed in Supplementary Table 1.
Within this subset of 40 couples (Fig. 2), 31 couples (Supplementary Table 1, ID: A1–A30, B1) were missing a parental sample from one partner. Through the use of sibling samples, successful haplotype analysis was achieved for 25 couples (80.6%, 25/31). However, haplotype analysis failed in 6 couples. For 2 of these couples, the haplotype was successfully constructed through single sperm sequencing, as the male partner’s parental samples were missing (Supplementary Fig. 4A). Additionally, three couples determined their haplotypes through reference embryos (Supplementary Fig. 4B). Unfortunately, haplotype analysis failed in 1 couple due to only one embryo achieved, in which the target gene was tested as normal, rendering it unsuitable as a reference embryo.

Haplotype analysis for 40 couples in Scenario III, classified by the number of missing parental DNA samples. Haplotypes were constructed using SNP sites from siblings’ DNA samples, single sperm, or affected embryos
Two couples lacked parental samples of the male partner (Fig. 2; Supplementary Table 1, ID: A31, B2). In one case, the male partner’s haplotype was successfully determined through SNP linkage analysis using samples from 5 siblings (Fig. 2; Supplementary Table 1, ID: A31). In the other case, paternal sample of the male partner was unavailable, and the female partner belonged to a heterozygous family. The two embryos obtained were found to possess the normal target gene, so it is unsuitable as a reference embryo (Fig. 2; Supplementary Table 1, ID: B2). Seven couples were unable to provide complete family samples from either side (Supplementary Table 1, ID: A32–A38). Ultimately, haplotypes were successfully analyzed in all 7 couples through the sibling samples or reference embryos.
For the 9 couples (ID: A26–A30, A33, A36, and B1–B2) whose haplotype analysis was unsuccessful despite the use of sibling samples, the analysis revealed two primary causes: (1) the siblings’ genotypes were consistent with the carriers, as observed in couples A30 and A33, and (2) the sibling genotypes were normal, and the parental genotypes provided were also normal, making it difficult to rule out the possibility of de novo mutations, as seen in couples A27–A29 (Supplementary Table 1).
For the additional 41 couples in Scenario III, who were unable to provide sibling samples, 40 couples successfully constructed the haplotype through reference embryos (Fig. 1). In one couple, both partners were devoid of paternal samples. The male partner’s haplotype was constructed via single sperm sequencing, while the female partner’s haplotype was accurately constructed through reference embryo analysis (Supplementary Fig. 4C).
In summary, haplotype linkage was successfully established in 99.4% (340/342) of couples. Parental haplotype analysis failed for two couples because the number of embryos obtained was low and the target gene in the embryos was normal, making them unsuitable for use as reference embryos for haplotype construction.
Laboratory test for embryos
Among the 342 couples included in the study, haplotype analysis ultimately failed for two couples with α-thalassemia, leading to the exclusion of three embryos from these two couples in the CNV analysis of embryos. Therefore, the study analyzed 1775 embryos from the remaining 340 couples.
(1) CNV results:
CNV detection is used to identify fragments with deletion/duplication ≥ 4 Mb, and mosaicism ≥ 30% of chromosome. The overall embryo amplification success rate was 97.7% (1734/1775). Of the amplified samples, 55.0% (953/1734) were euploid embryos, 32.9% (570/1734) were aneuploid embryos, and 12.2% (211/1734) were mosaic embryos (Table 2). Comparing the two WGA methods, the results showed that the MALBAC method outperformed the MDA method in terms of amplification success rate (98.8% vs. 96.2%), with a higher incidence of euploid embryos (57.9% vs. 50.6%, p = 0.003), and a reduced incidence of mosaicism (9.4% vs. 16.2%, p < 0.001) (Table 2).
(2) SNP linkage analysis and direct detection of target pathogenic mutation sites:
Due to the unavoidable occurrence of ADO during the WGA process, there is a certain risk of error in the direct detection of pathogenic loci in embryos. Therefore, the SNP linkage analysis method has been used simultaneously for more reliable results. It was worthwhile to investigate whether it was necessary to detect the target pathogenic mutation sites directly in WGA products derived from embryo biopsies.
Among the 340 couples, there were a total of 224 couples with α-thalassemia, accounting for 1194 embryos. Amplification failed in 32 (2.7%, 32/1194) embryos, and while amplification was successful, SNP linkage analysis failed in 42 (3.6%, 42/1162) embryos. Common factors for unsuccessful SNP linkage analysis included insufficient DNA samples or the number of available SNP sites. Thus, a total of 1120 embryos were included for analyzing the consistency between SNP linkage results and locus detection outcomes (Table 3). The success rate of detecting α-thalassemia loci was 96.3% (1078/1120), and the concordance between SNP linkage analysis results and locus detection results was 93.8% (1011/1078).
The study included 112 couples with beta-thalassemia, encompassing a total of 568 embryos. Nine (1.6%, 9/568) embryos failed amplification, and eleven (2.0%, 11/559) embryos exhibited successful amplification but had failed SNP linkage analysis. Consequently, 548 embryos were included for analyzing the consistency between SNP linkage results and sites detection results (Table 3). The success rate of detecting β-thalassemia loci was 100.0% (548/548), and the concordance between SNP linkage analysis results and locus detection results was 98.2% (538/548).
Additionally, among the four couples with α-/β-thalassemia, there were a total of 13 embryos. WGA amplification was successful for all embryos. The detection of the -α4.2 locus failed in two embryos. The locus detection results were also consistent with the analysis of the remaining 11 embryos for SNP linkage results, which was completed successfully.
Clinical outcomes
In this study, the PGT-M results were reported based on SNP linkage analysis results. A total of 256 couples, with an average maternal age of 31.4 ± 4.1 years, underwent embryo transfer guided by PGT-M combined with PGT-A. Three hundred two single blastocyst transfer cycles were tracked to clinical pregnancy follow-up. Among these cycles, one resulted in an ectopic pregnancy, with the remaining 301 cycles achieving a clinical pregnancy rate of 69.4% (209/301), which was close to the rate of 68.8% (11/16) reported by Chen et al. in their small-scale thalassemia PGT-M study [4]. Currently, 24 cycles have culminated in miscarriage, while 100 cycles have resulted in live births (Table 4). Notably, in 4 cycles, patients received the transfer of low-level mosaic embryos (excluding high-risk chromosomes) due to a lack of euploid embryos, with these transfers leading to 3 clinical pregnancies.
Of the remaining 86 couples, 51 did not have any viable embryos for transfer, accounting for 14.9% (51/342) of the total couples. The 35 couples have not undergone a transfer yet.
Amniocentesis results for 99 transfer cycles were collected, demonstrating a 100% concordance with the PGT-M results for the HBA and HBB genes.
To further validate the accuracy of the SNP linkage analysis, embryos with failed locus detection or inconsistent results with SNP linkage analysis were analyzed in relation to clinical outcomes. According to the results of CNV and SNP linkage analysis, 19 of the 42 embryos with successful SNP linkage analysis but unsuccessful locus detection in α-thalassemia (Table 3, Fig. 3) were eligible for transfer. Five embryos were transferred, resulting in four live births. Amniocentesis results were available for three patients, all of which were consistent with the SNP linkage analysis results (highlighted in orange in Supplementary Table 2).

Clinical outcomes for patients with successful embryo SNP analysis but discrepancies in mutation site detection versus SNP linkage analysis
There were 67 α-thalassemia embryos in which SNP linkage analysis was successful, but the mutation site testing results are inconsistent with the SNP linkage analysis (Table 3, Fig. 3). Among these, 34 embryos were identified as transferrable, and ultimately 10 embryos were transferred. Clinical pregnancies were established in six (60.0%, 6/10) of these cases. Amniocentesis was performed for three patients, with results corroborating the SNP linkage analysis (highlighted in dark green in Supplementary Table 2).
Discussion
This study was the first large-scale retrospective analysis of real-world PGT-M for α- and β-thalassemia, summarizing experiences in various patient pedigrees and offering a thorough and methodical framework for addressing various challenging scenarios encountered in PGT-M for thalassemia. Our findings suggest that using reference embryos for haplotype construction can significantly improve the success rate of PGT-M, potentially influencing future clinical guidelines. Due to the inevitable occurrence of ADO during WGA, there is a risk of misdiagnosis when directly testing pathogenic variant sites. Therefore, SNP linkage analysis becomes indispensable for the concurrent evaluation of embryos. Domestic guidelines further stipulate that PGT-M clinical testing should include both direct testing of gene pathogenic variant sites and linkage analysis of genetic polymorphic sites (such as STR or SNP), to mitigate diagnostic inaccuracies attributable to amplification failures and ADO [19]. Establishing parental haplotypes using the SNPs of probands or complete parental genotypes is a prerequisite for embryo SNP linkage analysis. However, in the real world, there were cases where couples were unable to provide samples from probands or complete parents for haplotype construction. In a previous study by Chen et al., involving PGT-M for thalassemia, when the probands or parental samples were unavailable, affected embryos were also used as reference embryos for haplotyping analysis [4]. The aim of that study was to establish a new method combining PGT-M and NGS, but the number of patients included was only 12, with the actual clinical application still unknown. In contrast, our extensive clinical study demonstrated 50 out of 342 couples (14.6%) successfully constructed haplotypes using reference embryos (Fig. 1), including cases of unavailable probands or complete parent samples, non-viable sibling samples, de novo mutations, or heterozygous pedigrees. Utilizing reference embryos for parental haplotype construction exhibited a notably high success rate.
In the study, haplotype analysis was unsuccessful for two out of the 342 couples due to having only one or two embryos available, all with normal target gene results, leaving no reference embryos. Consequently, these two couples need to proceed to the subsequent oocyte retrieval cycle for more embryos. For the male partner’s haplotype, the sequencing of single sperm could be an alternative option when family member samples were unavailable. This method had also been reported in PGT-M for other monogenic disorders [20]. In the study, the haplotype for the male partner was successfully achieved through single sperm sequencing in three other couples. Alternatively, third-generation sequencing may potentially be employed to determine haplotypes using the genomic DNA extracted from the couples’ peripheral blood samples.
At present, MALBAC and MDA represent the two principal methodologies for WGA. Liu et al.’s study showed that when the cell count reaches five or more, MALBAC and MDA exhibited similar detection efficiency and accuracy in PGT-M for β-thalassemia [16]. In the current study, both MALBAC and MDA demonstrated high amplification success rates for CNV analysis; however, MALBAC showed a statistically significantly higher amplification success rate compared to MDA (98.8% vs. 96.2%, p < 0.001). Additionally, MALBAC yielded a higher percentage of euploid embryos (57.9% vs. 50.6%, p = 0.003) (Table 2). The reason may be that MALBAC produces more dependable CNV results in single-cell sequencing than MDA because it has greater genome coverage, specificity, uniformity, and repeatability [16, 21, 22]. Compared to previous single-center studies, this multicenter analysis provides more robust data supporting the use of MALBAC for CNV analysis due to its higher amplification success rates.
The study provides data supporting for the accuracy and efficacy of NGS-based PGT-M. Patients were guided for embryo transfer based on the SNP linkage analysis and ploidy results in this study. Despite the successful application of SNP linkage analysis to 1668 embryos during practical embryo assessment, 119 embryos presented with either direct testing failures or results that were discordant with the SNP linkage analysis. This may be due to the limited number of cells in embryo biopsy, necessitating cell lysis and WGA. Although WGA offers high coverage of cell genome—MALBAC’s coverage can reach up to 93% [10, 22]—but there are still areas that cannot be randomly amplified. This is a common issue with all current WGA techniques. Additionally, WGA can introduce amplification bias, resulting in allelic dropout (ADO) at certain loci during the amplification process. Therefore, it is necessary to conduct both SNP linkage analysis and mutation site detection to ensure accuracy. When inconsistencies arise, the SNP linkage analysis results should be considered to guide the embryo transfer. Out of 256 patients who underwent single blastocyst transfer, 14.9% (51/342) had no transferable embryos. Among the patients who underwent embryo transfer and achieved ongoing pregnancies, 99 amniocentesis results were collected, revealing an absolute concordance (100%) between the HBA/HBB gene results in the amniotic fluid and the PGT-M results. Notably, 6 of the 99 patients had transferred embryos with either failed direct locus detection or inconsistent locus detection results compared to SNP linkage analysis results. The amniocentesis results showed complete consistency (100%) with the SNP linkage analysis results (Supplementary Table 2). An illustrative case from one family is delineated in Supplementary Table 3.
Additionally, the study acknowledges the low incidence of chromosomal segmental exchanges near to the pathogenic variant loci encountered in clinical settings. According to SNP linkage analysis, 0.6% of embryos (10 out of 1778) might have exhibited chromosomal exchanges near pathogenic variants. None of these embryos was transferred. Although the probability of chromosomal exchange is extremely low, it is advised to combine the SNP linkage analysis results and direct testing results of mutation sites to make a joint decision in such situations.
There are several strengths to this study. The study was the first large-scale retrospective analysis of real-world PGT-M for α- and β-thalassemia. Data were compiled from 342 couples across multiple centers between 2019 and 2022, encompassing amniocentesis results and clinical follow-ups. This extensive dataset was necessary to support the comprehensive and systematic analysis. Furthermore, the research was conducted from multiple aspects, including haplotype analysis methods, embryo testing techniques, and SNP linkage analysis results, to address the most challenging issues in current clinical practice. However, the study has a limitation. The study’s retrospective design and reliance on available samples may introduce selection bias. Future prospective clinical studies are needed to explore the accuracy of PGT-M.
In conclusion, this study was the first large-scale, real-world, multicenter retrospective clinical study on PGT-M for α- and β-thalassemia, yielding comprehensive recommendations for the clinical diagnosis of PGT-M for thalassemia. Sibling samples and reference embryos could be used to construct parental haplotype when traditional family samples are unavailable. When direct gene mutation detection fails or ADO occurs, embryo transfer can be considered based on SNP linkage analysis and ploidy results. This study’s comprehensive analysis offers valuable guidelines for enhancing the success of PGT-M for thalassemia.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–12.
Lai K, Huang G, Su L, He Y. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci Rep. 2017;7:920. https://doi.org/10.1038/s41598-017-00967-2.
Pan Y, Chen M, Zhang Y, Zhang M, Chen L, Lin N, et al. Analysis of genotype-phenotype correlation in patients with α-thalassemia from Fujian province, Southeastern China. J Clin Lab Anal. 2022;36:e24696. https://doi.org/10.1002/jcla.24696.
Chen D, Shen X, Wu C, Xu Y, Ding C, Zhang G, et al. Eleven healthy live births: a result of simultaneous preimplantation genetic testing of α- and β-double thalassemia and aneuploidy screening. J Assist Reprod Genet. 2020;37:549–57. https://doi.org/10.1007/s10815-020-01732-7.
Origa R. β-Thalassemia. Genet Med: Off J Am Coll Med Genet. 2017;19:609–19. https://doi.org/10.1038/gim.2016.173.
Muncie HL, Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80:339–44.
Casale M, Meloni A, Filosa A, Cuccia L, Caruso V, Palazzi G, et al. Multiparametric cardiac magnetic resonance survey in children with thalassemia major: a multicenter study. Circ Cardiovasc Imaging. 2015;8:e003230. https://doi.org/10.1161/circimaging.115.003230.
Kurtoglu AU, Kurtoglu E, Temizkan AK. Effect of iron overload on endocrinopathies in patients with beta-thalassaemia major and intermedia. Endokrynol Pol. 2012;63:260–3.
Huang X, Liu Y, Yu X, Huang Q, Lin C, Zeng J, et al. The clinical application of preimplantation genetic diagnosis for X-linked retinitis pigmentosa. J Assist Reprod Genet. 2019;36:989–94. https://doi.org/10.1007/s10815-019-01434-9.
Niu W, Wang L, Xu J, Li Y, Shi H, Li G, et al. Improved clinical outcomes of preimplantation genetic testing for aneuploidy using MALBAC-NGS compared with MDA-SNP array. BMC Pregnancy Childbirth. 2020;20:388. https://doi.org/10.1186/s12884-020-03082-9.
Xu H, Pu J, Wu Z, Huang Y, Han C, Li X. A healthy live birth after mosaic blastocyst transfer in preimplantation genetic testing for GATA1-related cytopenia combined with HLA matching. BMC Med Genomics. 2024;17:177. https://doi.org/10.1186/s12920-024-01951-2.
Mai AD, Harton GL, Quang VN, Van HN, Thi NH, Thuy NP, et al. Development and clinical application of a preimplantation genetic testing for monogenic disease (PGT-M) for beta thalassemia in Vietnam. J Assist Reprod Genet. 2021;38:365–74. https://doi.org/10.1007/s10815-020-02006-y.
Ou Z, Deng Y, Liang Y, Chen Z, Sun L. Using affected embryos to establish linkage phase in preimplantation genetic testing for thalassemia. Reprod Biol Endocrinol: RB&E. 2022;20:75. https://doi.org/10.1186/s12958-022-00948-9.
Wang Y, Zhu X, Yan Z, Zhi X, Guan S, Kuo Y, et al. Novel PGD strategy based on single sperm linkage analysis for carriers of single gene pathogenic variant and chromosome reciprocal translocation. J Assist Reprod Genet. 2020;37:1239–50. https://doi.org/10.1007/s10815-020-01753-2.
Gardner DK, Lane M, Stevens J, Schlenker T, Schoolcraft WB. Blastocyst score affects implantation and pregnancy outcome: towards a single blastocyst transfer. Fertil Steril. 2000;73:1155–8. https://doi.org/10.1016/s0015-0282(00)00518-5.
Liu W, Zhang H, Hu D, Lu S, Sun X. The performance of MALBAC and MDA methods in the identification of concurrent mutations and aneuploidy screening to diagnose beta-thalassaemia disorders at the single- and multiple-cell levels. J Clin Lab Anal. 2018; 32. https://doi.org/10.1002/jcla.22267.
Zhuang J, Tian J, Wei J, Zheng Y, Zhuang Q, Wang Y, et al. Molecular analysis of a large novel deletion causing α+-thalassemia. BMC Med Genet. 2019;20:74. https://doi.org/10.1186/s12881-019-0797-8.
Jomoui W, Panyasai S, Sripornsawan P, Tepakhan W. Revisiting and updating molecular epidemiology of α-thalassemia mutations in Thailand using MLPA and new multiplex gap-PCR for nine α-thalassemia deletion. Sci Rep. 2023;13:9850. https://doi.org/10.1038/s41598-023-36840-8.
Professional Committee on Reproductive Medicine CMDA. A Chinese experts’ consensus on preimplantation genetic testing for monogenic disorders. Chin J Reprod Contracep. 2021;41:477–85. https://doi.org/10.3760/cma.j.cn101441-20210118-00030.
Wang Y, Zhai F, Guan S, Yan Z, Zhu X, Kuo Y, et al. A comprehensive PGT-M strategy for ADPKD patients with de novo PKD1 mutations using affected embryo or gametes as proband. J Assist Reprod Genet. 2021;38:2425–34. https://doi.org/10.1007/s10815-021-02188-z.
Lu L, Lv B, Huang K, Xue Z, Zhu X, Fan G. Recent advances in preimplantation genetic diagnosis and screening. J Assist Reprod Genet. 2016;33:1129–34. https://doi.org/10.1007/s10815-016-0750-0.
Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science (New York, NY). 2012;338:1622–6. https://doi.org/10.1126/science.1229164.
Funding
The study was supported by the National Key R&D Program of China (2022YFC2703200), the Shenzhen Municipal Science and Technology Innovation Committee (Multi-party Collaboration Project) Key Project for Basic Research (JCYJ20200109140623124), the National Natural Science Foundation of China (82160028), and the Guangxi Key Laboratory of reproductive health and birth defect prevention (21-220-22).
Author information
Authors and Affiliations
Contributions
ZR, PH, and YW conceived and designed the experiments. LNX, JHS, LZ, and XLL performed the experiments and analyzed the data of the work. ZQZ, CHZ, BLS, and DMZ conducted the collection and analysis of clinical data. JR and YXY helped to revise the manuscript. CF, HZ, WPQ, and SJL supervised and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the relevant committees at each participating center (No. 2024ZSLYEC-323). Each couple provided written informed consent prior to the PGT cycle, and if peripheral blood samples from other family members were required, additional informed consent was obtained. Due to the retrospective design of the study, there was no need for extra participant declaration consent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, Z., Huang, P., Wang, Y. et al. Technically feasible solutions to challenges in preimplantation genetic testing for thalassemia: experiences of multiple centers between 2019 and 2022. J Assist Reprod Genet (2024). https://doi.org/10.1007/s10815-024-03240-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10815-024-03240-4