
Abstract
Purpose
To evaluate the efficacy of preimplantation genetic testing (PGT) for α- and β-double thalassemia combined with aneuploidy screening using next-generation sequencing (NGS).
Methods
An NGS-based PGT protocol was performed between 2017 and 2018 for twelve couples, each of which carried both α- and β-thalassemia mutations. Trophectoderm biopsy samples underwent whole-genome amplification using multiple displacement amplification (MDA), followed by NGS for thalassemia detection and aneuploidy screening. A selection of several informative single nucleotide polymorphisms (SNPs) established haplotypes. Aneuploidy screening was performed only on unaffected noncarriers and carriers. Unaffected and euploid embryos were transferred into the uterus through frozen-thawed embryo transfer (FET).
Results
A total of 280 oocytes were retrieved following 18 ovum pick-up (OPU) cycles, with 182 normally fertilized and 112 cultured to become blastocysts. One hundred and seven (95.5%, 107/112) blastocysts received conclusive PGT results, showing 56 (52.3%, 56/107) were unaffected. Thirty-seven (66.1%, 37/56) of the unaffected were also identified as euploid. One family had no transferable embryos. Unaffected and euploid embryos were then transferred into the uterus of the other 11 couples resulting in 11 healthy live births. The clinical pregnancy rate was 61.1% (11/18) per OPU and 68.8% (11/16) per FET, with no miscarriage reported. Seven families accepted the prenatal diagnosis and received consistent results with the NGS-based PGT.
Conclusion
This study indicated that NGS could realize the simultaneous PGT of double thalassemia and aneuploidy screening in a reliable and accurate manner. Moreover, it eliminated the need for multiple biopsies, alleviating the potential damages to the pre-implanted blastocysts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thalassemia is a group of hereditary anemias characterized by reduced or even absence of one of the globin chains of hemoglobin (Hb), especially prevalent in the Mediterranean region and Southeast Asia. As of 2017, it occurs in about 298.6 million people in the world [1]. In the Chinese mainland, the prevalence of α-thalassemia and β-thalassemia ranged from 1.20~19.87% and 0.53~6.84%, respectively, with the highest prevalence found in Guangxi and Guangdong province [2].
The α-globin gene cluster is located on chromosome 16p13.3 and comprised one embryonic ξ-globin gene and two α-globin genes, α2 and α1, in tandem (in cis) [3]. Southeast Asia deletion (--SEA) is the most common homozygous mutation of α-thalassemia in China with a gene frequency of 2.54% [2]. Homozygotes with Southeast Asia deletion suffer from Hb Bart’s hydrops fetalis syndrome and usually die either in late gestation or within a few minutes after birth [4].
β-Thalassemia is a group of hereditary blood disorders characterized by reduced (β+) or absent (β0) β-globin chain synthesis, resulting in reduced Hb levels in red blood cells (RBC), decreased RBC production, and anemia. They are caused by point mutations or, more rarely, deletions in the β-globin gene cluster on chromosome 11 [3]. Infants with severe β-thalassemia are usually diagnosed before 2 years old and require regular RBC transfusions to survive [5].
Prenatal diagnosis is advocated in China to prevent the birth of severe thalassemia babies. However, it is an invasive procedure that may induce infection, miscarriage, and the torture of terminating affected pregnancy. As an alternative to prenatal diagnosis, preimplantation genetic testing (PGT) for monogenic diseases (PGT-M) makes it possible to diagnose before the initiation of pregnancy and precludes the need to terminate affected pregnancies. PGT-M has been successfully applied for the detection of α-thalassemia or β-thalassemia, with only two reports of simultaneous detection of both α- and β-thalassemias of embryos. Previously, our team has successfully achieved simultaneous PGT-M of both α- and β-thalassemia by multiple displacement amplification (MDA) combined with short tandem repeat (STR) haplotyping and allele-specific amplification [6, 7]. However, these methods have their shortcomings: the PCR-based method is adversely influenced by allele drop-outs (ADOs), and STR markers tend to be semi-informative [8], limiting their widespread use in PGT-M.
Next-generation sequencing (NGS) is the latest breakthrough for PGT-M and PGT for aneuploidy (PGT-A), in merits of reliability, higher throughput, and personalized assays. Analysis of single nucleotide polymorphisms (SNPs) linked to causative gene regions with NGS technology enables an all-in-one genetic and chromosomal testing [9]. In this study, we carried out the simultaneous diagnosis of both α- and β- thalassemias combined with aneuploidy screening using an MDA-NGS based PGT method.
Materials and methods
Patients
Twelve couples came to our center asking for PGT treatment between 2017 and 2018, as each of them carried both α- and β-thalassemia mutations. The patients’ characteristics, including age, mutation types, and reproductive history, were shown in Table 1. Written consents were obtained from all the families. The study was approved by the Research Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University.
Pedigree analysis
Genotyping data of SNPs of the parents and their relatives or affected fetus were determined for haplotyping. Informative SNPs closely related with the mutated causative gene were used to deduce the pathogenicity of the embryos. When the DNA of their relatives or affected fetus were not available, pedigree analysis was performed through directly detecting mutation sites of the embryos with NGS and using the affected embryos as probands.
Ovarian stimulation and ICSI procedure
Ovarian stimulation, intracytoplasmic sperm injection procedure, and embryo culture were carried out as previously reported [10].
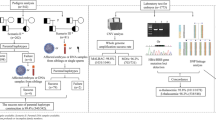
Embryo biopsy
Fresh or frozen-thawed blastocysts with at least average grade (Gardner & Schoolcraft, 1999) were firstly created a hole in the zona pellucida with a noncontact laser on the morning of day 5 or day 6. The time between laser-assisted zona opening and biopsy was 3–5 h. Only blastocysts with multiple trophectoderm cells herniating from the zona pellucida were considered eligible for biopsy. Five to ten trophectoderm cells were aspirated using a biopsy pipette with an inner diameter of 30 μm, and a dissection was performed using the OctaxShot™ laser system. Blastocysts were vitrified immediately after the biopsy using a Kitazato vitrification kit (Kitazato Biopharma Co., Ltd.). Online resource 1 displays the photos of the embryo biopsy procedure.
Whole-genome amplification and next-generation sequencing
The biopsied trophectoderm cells transferred to lysis buffer were subjected to whole-genome amplification (WGA) using the MDA approach (REPLI-g Single Cell Kit, QIAGEN Inc.). The MDA products were used to perform NGS (Ion Torrent™) for PGT-M of α- and β-thalassemia and PGT-A.
We selected the SEA region, including HBA2 gene (NM_000517.4 chr16: 223,300 to 227,103) and HBA1 gene (NM_000558.4 chr16:215400–234,700), as the target region for α-thalassemia. Also, the HBB gene (NM_000518.4 chr11:5246696–5,248,301), combined with its flanking 10-bp sequence, was selected as the target region for β-thalassemia. A series of SNP loci upstream and downstream of the target regions were selected as genetic markers (Table 3). We employed semiconductor sequencing using the Ion Torrent Personal Genome Machine to detect the genotypes of SNPs, and the informative SNPs were selected to construct haplotypes for linkage analysis.
All primers required for NGS were designed through the ION AMPLISEQ™ DESIGNER website.
We performed PGT-A only for unaffected embryos, including unaffected noncarrier and carrier embryos.
Embryo transfer
Blastocysts diagnosed as unaffected and euploid were identified as transferrable embryos, which were thawed and transferred into the uterus in subsequent frozen-thawed embryo transfer (FET) cycles through standard procedures.
Results
The twelve couples experienced a total of 18 ovum pick-up (OPU) cycles and 12 PGT cycles. Two hundred eighty oocytes were retrieved, among which 182 were normally fertilized with ICSI method and 112 cultured to become blastocysts. Trophectoderm biopsy and NGS were performed on the 112 blastocysts. One hundred and seven (95.5%, 107/112) blastocysts received conclusive PGT-M results. As the results of α-thalassemia were obtained faster than those of β-thalassemia using our NGS-based PGT, we did not conduct testing of β-thalassemia for family 4 when we found that all their embryos were abnormal in the testing of α-thalassemia. Fifty-six (52.3%, 56/107) of the embryos were found to be unaffected for both α- and β-thalassemias (Table 2). PGT-A results of these 56 embryos showed that 18 were aneuploid, and one was undetectable. Online resource 2 presents the detailed PGT results of the 112 embryos.
Therefore, 37 (66.1%, 37/56) embryos diagnosed as unaffected and euploid were considered suitable for transfer. One family had no transferrable embryos. For the other 11 couples, each family went through 1–3 FET cycles, with one embryo transferred per cycle. The clinical pregnancy rate was 61.1% (11/18) per OPU cycle and 68.8% (11/16) per FET cycle. All 11 families achieved healthy live births with no miscarriage observed. Seven families accepted the prenatal diagnosis and showed consistent results with the NGS-based PGT-M/PGT-A. Family 11, who only accepted the prenatal diagnosis for thalassemia detection, also achieved the same result with PGT-M (Table 2).
Take family 1 as an example (the pathogenic genotype and reproductive history are shown in Table 1). Their theoretical probability of having children with a normal genetic phenotype is 37.5%, which is a definite indication of PGT-M. As II-2 is an HbH patient, and II-1 is a heterozygote for -α3.7 deletion, according to the Hereditary Law, their embryos formed will have a 50% chance of suffering HBH disease and a 50% chance of being α-thalassemia carriers (Fig. 1). Therefore, during the haplotype analysis of α- thalassemia, the transferrable embryo should be the one that inherits the unaffected haplotype of II-1. Since the DNA samples from their aborted fetuses were not available, we detected α-thalassemia on the parents of II-1 and found that his father (I-1) was also a heterozygote of -α3.7 deletion, while his mother (I-2) had a normal genotype. Therefore, we believed that the affected α- thalassemia haplotype of II-1 was derived from I-1 (Fig. 1).

Pedigree analysis of α-thalassemia (family 1). The deep red background indicates the paternal affected haplotype (-α3.7). The bright red background indicates one of the maternal affected haplotypes (--SEA). The pink background indicates the other maternal affected haplotype (ααcs)
Next, we performed NGS on the WGA products of I-1, I-2, II-1, II-2, and the ten embryos for simultaneous mutation detection and SNP-based haplotype analysis of α- and β-thalassemias (Figs. 2 and 3). Chromosome aneuploidy was screened in the meantime, indicating that three of the ten embryos were aneuploid. (Online Resource 2). In the end, only one of the ten embryos (E-1) was a carrier of α-thalassemia (the remaining nine were diagnosed with HbH disease), and a heterozygote for CD17 mutation, and its PGT-A result was euploid. After learning the diagnosis, this couple decided to transfer E-1, resulting in a singleton pregnancy. Prenatal diagnosis of thalassemia and chromosome abnormalities was carried out at 4+ months of pregnancy through amniocentesis, showing consistent results with NGS-PGT.

Part of the α-thalassemia haplotype results by SNP (family 1). Position 22597 is the maternal mutation site of ααcs. Between the solid horizontal lines is the area of the SEA deletion. Between the double horizontal lines is the area of -α3.7 deletion. The deep red background indicates the paternal affected haplotype (-α3.7). The bright red background indicates one of the maternal affected haplotypes (--SEA). The pink background indicates the other maternal affected haplotype (ααcs). “?” represents that the site is not detected

Part of the β-thalassemia haplotype results by SNP (family 1). Position 5248200 is the maternal mutation site of CD17, which is also marked in bright purple. Between the solid horizontal lines is the area of the HBB gene. The deep red background indicates the paternal affected haplotype. The bright red background indicates the maternal affected haplotype
Discussion
In this study, we performed simultaneous PGT of α- and β-thalassemia combined with aneuploidy screening on 112 embryos for twelve couples, resulting in ideal diagnostic results and 11 healthy live births, which demonstrated the great versatility, reliability, and safety of NGS-based PGT.
Previously, simultaneous PGT-M of both α- and β-thalassemia was successfully achieved in our center using MDA combined with STR haplotyping and PCR-based mutation detection [6, 7]. Moreover, Kakourou et al. used PCR-based protocol combining with STR linkage analysis to apply PGT-M for β-thalassemia and sideroblastic anemia [11]. However, the PCR-based method is inevitably interfered with ADOs. Though haplotype analysis with STRs may reduce the effect of ADO, the number of STR loci is limited, with some of them tend to be semi-informative. Besides, the recombination between STRs and target genes may affect the diagnosis efficiency [8, 12]. These suggest that the methods of the previous studies could be further improved.
In PGT-M cycles, thalassemia patients of advanced maternal age or with a history of recurrent spontaneous abortion may require spontaneous aneuploidy screening. Although PGT-A had a controversial reputation in the past [13,14,15], the second generation of PGT-A based on biopsy of blastocyst plus comprehensive chromosome screening has been proved to have promising clinical effect in recent years [16,17,18,19]. A study of Rechitsky et al. showed a statistically increased pregnancy rate (68.4% vs. 45.4%) and a 3-fold reduction of spontaneous abortion rate (15% vs. 5.5%) in PGT-M plus PGT-A cycles compared with PGT-M alone cycles [20]. Similarly, in our study, family 9 and 10 had a history of early embryonic death or fetal chromosomal abnormality. Combining with PGT-A screening, their euploid and unaffected embryos were successfully implanted and resulted in healthy live births. Moreover, a recent study from our center demonstrated that even for women at a younger age, PGT-M plus PGT-A significantly improved the live birth rate in their first FET cycles compared with PGT-M alone (61.22% vs. 43.98%) [21]. Therefore, it is fascinating to supply additional PGT-A to PGT-M. Admittedly, PGT-A aggravates the financial burden and has not yet shown superiority in increasing the cumulative live birth rate [22]. To save costs for patients, we only performed additional PGT-A for embryos diagnosed as unaffected in PGT-M. And recently, a theoretical cost-effectiveness study confirmed a notion that PGT-A would result in higher cost-effectiveness with an increase of maternal age [23].
Compared with previous PGT-M methods, such as PCR and STR, NGS-based SNP haplotyping could reduce misdiagnosis by linkage analyses with multiple SNP loci. It has been successfully applied to the detection of HbH disease, primary open angle glaucoma, and PKD2 gene mutations on human preimplantation embryos, indicating the high accuracy, fidelity, and consistency with Sanger sequencing [24,25,26]. Moreover, our results support that NGS-based method allows the simultaneous detection of aneuploidy, targeted mutation sites, and their linked SNPs in a one-step procedure, making it possible to provide multiple diagnosis results in a single PGT cycle. In our study, 32.1% (37/56) embryos unaffected from thalassemias were diagnosed as aneuploid. As aneuploidy is one of the essential causes of spontaneous abortion [27], this result supports the superiority of NGS in the integration of genetic disease diagnosis and aneuploidy screening. Similar to our study, Backenroth et al. developed an all-in-one NGS-based workflow for preimplantation molecular and chromosomal diagnosis, showing the valuable application prospects of NGS in the field of PGT [9].
Compared with karyomapping technology, another advantage of NGS-based haplotyping is that when absent of affected relatives in the family, haplotyping can still be performed through directly detecting mutation sites with NGS and using affected embryos or gametes as probands [28, 29]. Our study further supports this finding as we achieved ideal haplotyping analysis for five couples (families 7, 8, 9, 11, 12) under this approach.
There have been several reports of simultaneous detection of both α- and β-thalassemias with a relatively large amount of DNA samples, such as blood, amniotic fluid, chorionic villus, or other genomic DNA (gDNA) samples [30,31,32]. However, for embryonic samples, the DNA content is in the picogram level, which brings a considerable challenge for multipurpose PGT. For this reason, biopsies used to be performed twice at different stages of embryo development, cleavage stage, and blastocyst stage, to diagnose two genetic diseases or to combine mutation detection with aneuploidy screening. However, multiple biopsies have been proved to decrease embryo potentials [33]. Recently, with the rapid development of genome amplification and detection technologies, several studies have demonstrated that multipurpose PGT can be realized with a single biopsy [6, 9, 11, 34]. Similarly, using the NGS-based protocol of our study, only one blastocyst biopsy was desired to achieve both double thalassemia detection and aneuploidy screening, which alleviated the negative effect of multiple biopsies on embryos.
The genotypes of the couple determine the theoretical probability of their embryos unaffected from both α- and β-thalassemias. When the couple is double heterozygotes of α- and β-thalassemia, the probability is 0.75 × 0.75 = 56.25%; our results showed that the probability was 59.8% (52/87), close to the theoretical value. When one partner is HbH patients (such as family 1, 5, and 10), the probability is 0.5 × 0.75 = 37.5%; the probability was 20% (4/20) in our study, lower than the theoretical value, probably due to the small sample size. Notably, the probability of obtaining a transferable embryo will be declined when a PGT-A diagnosis needs to be combined. For example, the probability was reduced to 37.9% (33/87) in our study for the couples who were double heterozygotes of α- and β-thalassemia. According to our previous study, we believe that at least four biopsied embryos are required to obtain at least one unaffected embryo in a PGT-M cycle under the condition of basal FSH level smaller than 8.0 mmol/L [35]. Undoubtedly, our PGT protocol, which combines double thalassemia detection and chromosome screening, requires an even more significant number of biopsied embryos to obtain at least one transferrable. Some families could not get enough blastocysts for diagnosis in one OPU cycle, making them enter several ovarian stimulation cycles to accumulate more. For example, family 4 experienced three hyper-stimulation cycles; for families 5, 6, 9, and 12, each family went through two OPU cycles (Table 3).
Conclusion
To our knowledge, this is the largest study of simultaneous PGT for α- and β-double thalassemia combined with aneuploid screening under the NGS approach. Our results indicated that NGS could be a reliable method enabling simultaneous PGT for multiple purposes, which would improve the clinical value of a single PGT cycle. Moreover, it eliminated the need for multiple biopsies, which alleviated potential damage to the pre-implanted blastocysts.
References
Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–858.
Lai K, Huang G, Su L, He Y. The prevalence of thalassemia in mainland China: evidence from epidemiological surveys. Sci Rep. 2017;7(1):920.
Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2018;32(2):177–91.
Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–67.
Origa R. beta-Thalassemia. Genet Med. 2017;19(6):609–19.
Shen XT, Xu YW, Zhong YP, Zeng YH, Wang J, Ding CH, et al. Combination of multiple displacement amplification with short tandem repeat polymorphismin preimplantation genetic diagnosis. Beijing Da Xue Xue Bao Yi Xue Ban. 2013;45(6):852–8.
Shen X, Xu Y, Zhong Y, Zhou C, Zeng Y, Zhuang G, et al. Preimplantation genetic diagnosis for alpha-and beta-double thalassemia. J Assist Reprod Genet. 2011;28(10):957–64.
Natesan SA, Bladon AJ, Coskun S, Qubbaj W, Prates R, Munne S, et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet Med. 2014;16(11):838–45.
Backenroth D, Zahdeh F, Kling Y, Peretz A, Rosen T, Kort D, et al. Haploseek: a 24-hour all-in-one method for preimplantation genetic diagnosis (PGD) of monogenic disease and aneuploidy. Genet Med. 2018.
Xu YW, Zeng YH, Deng J, Liu Y, Gao L, Zhou CQ, et al. Preimplantation genetic diagnosis for alpha-thalassaemia in China. J Assist Reprod Genet. 2009;26(7):399–403.
Kakourou G, Vrettou C, Kattamis A, Destouni A, Poulou M, Moutafi M, et al. Complex preimplantation genetic diagnosis for beta-thalassaemia, sideroblastic anaemia, and human leukocyte antigen (HLA)-typing. Syst Biol Reprod Med. 2016;62(1):69–76.
Gueye NA, Jalas C, Tao X, Taylor D, Scott RT Jr, Treff NR. Improved sensitivity to detect recombination using qPCR for Dyskeratosis Congenita PGD. J Assist Reprod Genet. 2014;31(9):1227–30.
Mastenbroek S, Twisk M, van Echten-Arends J, Sikkema-Raddatz B, Korevaar JC, Verhoeve HR, et al. In vitro fertilization with preimplantation genetic screening. N Engl J Med. 2007;357(1):9–17.
Hardarson T, Hanson C, Lundin K, Hillensjo T, Nilsson L, Stevic J, et al. Preimplantation genetic screening in women of advanced maternal age caused a decrease in clinical pregnancy rate: a randomized controlled trial. Hum Reprod. 2008;23(12):2806–12.
Blockeel C, Schutyser V, De Vos A, Verpoest W, De Vos M, Staessen C, et al. Prospectively randomized controlled trial of PGS in IVF/ICSI patients with poor implantation. Reprod BioMed Online. 2008;17(6):848–54.
Scott RT Jr, Upham KM, Forman EJ, Hong KH, Scott KL, Taylor D, et al. Blastocyst biopsy with comprehensive chromosome screening and fresh embryo transfer significantly increases in vitro fertilization implantation and delivery rates: a randomized controlled trial. Fertil Steril. 2013;100(3):697–703.
Forman EJ, Hong KH, Ferry KM, Tao X, Taylor D, Levy B, et al. In vitro fertilization with single euploid blastocyst transfer: a randomized controlled trial. Fertil Steril. 2013;100(1):100–107.e101.
Gleicher N, Barad DH. A review of, and commentary on, the ongoing second clinical introduction of preimplantation genetic screening (PGS) to routine IVF practice. J Assist Reprod Genet. 2012;29(11):1159–66.
Lee CI, Wu CH, Pai YP, Chang YJ, Chen CI, Lee TH, et al. Performance of preimplantation genetic testing for aneuploidy in IVF cycles for patients with advanced maternal age, repeat implantation failure, and idiopathic recurrent miscarriage. Taiwan J Obstet Gynecol. 2019;58(2):239–43.
Rechitsky S, Pakhalchuk T, San Ramos G, Goodman A, Zlatopolsky Z, Kuliev A. First systematic experience of preimplantation genetic diagnosis for single-gene disorders, and/or preimplantation human leukocyte antigen typing, combined with 24-chromosome aneuploidy testing. Fertil Steril. 2015;103(2):503–12.
Hou W, Xu Y, Li R, Song J, Wang J, Zeng Y, et al. Role of aneuploidy screening in preimplantation genetic testing for monogenic diseases in young women. Fertil Steril. 2019;111(5):928–35.
Rubio C, Bellver J, Rodrigo L, Castillon G, Guillen A, Vidal C, et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: a randomized, controlled study. Fertil Steril. 2017;107(5):1122–9.
Somigliana E, Busnelli A, Paffoni A, Vigano P, Riccaboni A, Rubio C, et al. Cost-effectiveness of preimplantation genetic testing for aneuploidies. Fertil Steril. 2019;111(6):1169–76.
Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. The clinical application of NGS-based SNP haplotyping for PGD of Hb H disease. Syst Biol Reprod Med. 2017;63(3):212–7.
Ji X, Zhang Z, Shi J, He B. Clinical application of NGS-based SNP haplotyping for the preimplantation genetic diagnosis of primary open angle glaucoma. Syst Biol Reprod Med. 2019;65(3):258–63.
Chen SC, Xu XL, Zhang JY, Ding GL, Jin L, Liu B, et al. Identification of PKD2 mutations in human preimplantation embryos in vitro using a combination of targeted next-generation sequencing and targeted haplotyping. Sci Rep. 2016;6:25488.
Jia CW, Wang L, Lan YL, Song R, Zhou LY, Yu L, et al. Aneuploidy in early miscarriage and its related factors. Chin Med J. 2015;128(20):2772–6.
Chen L, Diao Z, Xu Z, Zhou J, Yan G, Sun H. The clinical application of single-sperm-based SNP haplotyping for PGD of osteogenesis imperfecta. Syst Biol Reprod Med. 2019;65(1):75–80.
Ren Y, Zhi X, Zhu X, Huang J, Lian Y, Li R, et al. Clinical applications of MARSALA for preimplantation genetic diagnosis of spinal muscular atrophy. J Genet Genomics. 2016;43(9):541–7.
Lin M, Zhu JJ, Wang Q, Xie LX, Lu M, Wang JL, et al. Development and evaluation of a reverse dot blot assay for the simultaneous detection of common alpha and beta thalassemia in Chinese. Blood Cells Mol Dis. 2012;48(2):86–90.
Bang-Ce Y, Hongqiong L, Zhuanfong Z, Zhengsong L, Jianling G. Simultaneous detection of alpha-thalassemia and beta-thalassemia by oligonucleotide microarray. Haematologica. 2004;89(8):1010–2.
Siriratmanawong N, Fucharoen G, Sanchaisuriya K, Ratanasiri T, Fucharoen S. Simultaneous PCR detection of beta-thalassemia and alpha-thalassemia 1 (SEA type) in prenatal diagnosis of complex thalassemia syndrome. Clin Biochem. 2001;34(5):377–80.
Haapaniemi Kouru K, Malmgren H, Nordenskjold M, Fridstrom M, Csemiczky G, Blennow E. One-cell biopsy significantly improves the outcome of preimplantation genetic diagnosis (PGD) treatment: retrospective analysis of 569 PGD cycles at the Stockholm PGD centre. Hum Reprod. 2012;27(9):2843–9.
Minasi MG, Fiorentino F, Ruberti A, Biricik A, Cursio E, Cotroneo E, et al. Genetic diseases and aneuploidies can be detected with a single blastocyst biopsy: a successful clinical approach. Hum Reprod. 2017;32(8):1770–7.
Hu X, Wang J, Li Y, Wang Y, Ding C, Zeng Y, et al. Clinical considerations of preimplantation genetic diagnosis for monogenic diseases. PLoS One. 2015;10(9):e0139613.
Acknowledgments
The authors are grateful for the support from the Guangdong Provincial Key Laboratory of Reproductive Medicine (2012A061400003) and Guangzhou Science and Technology Project: Development and Application of The Reagents for Preimplantation Diagnosis and Screening by Next-Generation Sequencing (201704020217).
Author information
Authors and Affiliations
Contributions
D.C. and X.S. designed and performed the experiments, collected and analyzed data, and wrote the manuscript. C.W., Y.X., and C.D. conducted the experiment and contributed to the interpretation of the results. C.Z., Y-W.X., and G.Z. conceived, designed, supervised the experiments, and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval was obtained from the Research Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University. A detailed written consent was obtained from each family for being included in this study.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dongjia Chen and Xiaoting Shen are the joint first author
Rights and permissions
About this article

Cite this article
Chen, D., Shen, X., Wu, C. et al. Eleven healthy live births: a result of simultaneous preimplantation genetic testing of α- and β-double thalassemia and aneuploidy screening. J Assist Reprod Genet 37, 549–557 (2020). https://doi.org/10.1007/s10815-020-01732-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-020-01732-7