
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited kidney disease characterized by the development of renal cysts and progression to renal failure. Preimplantation genetic testing-monogenic disease (PGT-M) is an alternative option to obtain healthy babies. However, de novo PKD1 mutation of one of the spouses or the absence of a positive family history poses a serious challenge to PGT-M. Here, we described a comprehensive strategy which includes preimplantation genetic testing for aneuploidies (PGT-A) study and monogenic diagnosis study for ADPKD patients bearing de novo mutations. The innovation of our strategy is to use the gamete (polar body or single sperm) as proband for single-nucleotide polymorphism (SNP) linkage analysis to detect an embryo’s carrier status. Nine ADPKD couples with either de novo mutation or without a positive family history were recruited and a total of 34 embryos from 13 PGT-M cycles were examined. Within these nine couples, two successfully delivered healthy babies had their genetic status confirmed by amniocentesis. This study provides a creative approach for embryo diagnosis of patients with de novo mutations or patients who lack essential family members for linkage analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a common single-gene kidney disease with an incidence rate of 1/1000–1/400 [1]. ADPKD patients mainly show the progressive growth of renal cysts accompanied by hypertension [2], kidney stones [3], renal pain, hematuria [4], and mild proteinuria [5], resulting in end-stage renal disease (ESRD) and renal failure in adulthood.
ADPKD is mainly caused by two single-gene mutations: PKD1 (polycystin-1, PC1) located at 16p13.3 [6, 7] or PKD2 (polycystin-2, PC2) located at 4q21-23 [8]. Dysfunction of either protein will cause the primary cilia to function abnormally, leading to a series of cell signal transduction changes and abnormal cell proliferation and secretion that result in the formation and progressive enlargement of cysts [9]. About 85% of ADPKD patients carry PKD1 mutation and the rest carry PKD2 mutation [6, 7]. There are six pseudogenes 13 to 16 megabases away from PKD1 and the homology between pseudogenes and the duplicated regions of PKD1 is up to 97.7% [10]. In addition to the complex gene structure, the GC content of PKD1 is as high as 70 to 80% [11]. During amplification, the homologous pseudogenes and high GC content interfere with the identification of true PKD1 mutations.
At present, there is no effective treatment for ADPKD patients. Hence, preventing the transmission of mutated PKD1 gene to their offspring is of great importance. PGT-M (preimplantation genetic testing for monogenic defects) in assisted reproductive technology is an effective way to block disease inheritance. During in vitro fertilization (IVF) cycle, blastocyst biopsy and subsequent genetic diagnosis can detect the carrier status of embryos. In this way, the detected unaffected embryos will be transferred. It has been reported that two healthy babies were born through PGD based on short tandem repeat (STR) linkage analyses following blastomere biopsy [12]. Verlinsky et al. [13] used polar bodies and blastomere biopsies for subsequent STR linkage analyses to examine the embryo for maternal mutations. These methods were only applicable to couples with a definite proband and a complete medical family history for linkage analyses. However, due to de novo mutation, 10–25% of ADPKD patients do not have an informative family history, thus hindering proper diagnosis [14]. For male de novo mutation, Shi et al. [15] reported a strategy based on single sperms for linkage analyses. Nevertheless, the previous methods failed to cover all de novo mutation patients, especially for females. Here, we developed a comprehensive strategy using gamete or embryo as proband for linkage analyses (GEPLA) that could be applied to all patients. GEPLA strategy uses affected embryo as proband (EAP) or gametes carrying mutant alleles as proband (GAP) for SNP linkage analyses. Embryo carriers identified through direct mutation detection were preferentially taken as probands for subsequent haplotype analyses. For couples without EAP, single sperms or the second polar body (PB2) was complementally selected to identify mutant alleles, which could be regarded as proband for the following haplotype analyses.
In this study, we recruited nine couples without positive ADPKD family history. Detailed clinical information was collected by researchers and clinicians at our center. Specific diagnostic strategies were designed for these patients. Finally, all couples accepted IVF-PGT treatment and four unaffected embryos were transferred. As a result, two healthy babies were born whose carrier statuses were confirmed by amniotic analysis. This comprehensive strategy indicates the accuracy and universality of embryo diagnosis for patients who have de novo mutations or lack a useful family history.
Materials and methods
Patients
From 2016 to 2020, nine couples were recruited and authorized to be treated in our de novo PGT-M study for polycystic kidney disease at the Reproductive Center of Peking University Third Hospital. The clinical diagnosis of ADPKD was based on the number of kidney cysts detected by ultrasonography, as described in unified criteria by Pei et al. [16] All patients included in the study met the diagnostic criteria for ADPKD and have no affected family members. Genetic counseling and pre-validation tests suggested that all nine ADPKD patients had de novo pathogenic PKD1 mutations. Pedigree charts of all these cases were shown in Supplemental Figure 1. Among these nine cases, eight were males and one was female (case 2). The basic information for these nine couples is shown in Table 1. This study was approved by Peking University Third Hospital and informed consents were signed by all study participants.
In vitro fertilization (IVF) and single-sperm collection
All patients received in vitro fertilization and embryo transfer (IVF-ET) treatment. As shown in Table 1, couple 4 and couple 6 received 2 and 3 cycles of IVF-ET respectively, while other couples received 1 IVF-ET cycle. A total of 108 matured metaphase II (MII) stage oocytes were collected and fertilized through intracytoplasmic sperm injection (ICSI); 34 embryos reached the embryo biopsy standards. Unfortunately, for couple 4, no embryos reached the embryo biopsy standards. After ICSI, a certain amount of single sperms was collected from each male patient (except for couple 2) as backups for subsequent diagnosis.
Semen quality was firstly assessed by sperm concentration, motility, and morphology. Higher motile sperms (n = 10) with morphological integrity were captured and collected individually into ten separate PCR tubes [17]. Following the flexible strategy (Fig. 5), if no EAP was found, single sperms would come in handy. After whole-genome amplification (WGA), a portion of the WGA products was used for mutant allele identification by specific PCR and Sanger sequencing. The rest of the WGA products from single sperm as proband (SAP) were used for NGS and haplotype analyses.
Biopsy and polar body collection
For all cases, blastocyst biopsy was applied. At day 5 or day 6 after fertilization, trophectoderm (TE) biopsy of the hatched blastocysts was performed. The biopsied TE cells were washed with PBS and then kept in the lysis buffer (5 μL) in a 0.2-mL PCR tube. After the biopsy, the blastocysts were frozen by vitrification.
For couple 2, the only female patient, an additional polar body biopsy was performed prior to blastocyst biopsy. PB2 were obtained just after ICSI and 8–14 h after fertilization and put into PCR tubes [18]. Similar to the single-sperm backups, PB2 is an additional option if no EAP was identified. After whole-genome amplification (WGA), the WGA products were used for mutant allele analyses by specific PCR, Sanger sequencing, and haplotype analyses based on NGS data.
WGA and direct mutation detection
Whole-genome DNA from lysed cells was amplified using the commercial MALBAC amplification kit (Yikon Genomics Inc., China). MALBAC products were then reamplified using specific primers targeting the mutation sites. The primers were designed to specifically amplify the PKD1 mutation and to have amplicons containing at least one different site to distinguish PKD1 from the pseudogenes. Touchdown PCR was used to ensure the specific amplification of PKD1 targeted sequences and was performed at 98 °C for 30 s, 30 cycles of 15 s at 98 °C, 30 s at 65–55 °C (annealing temperature from 65 to 55°C and 0.5 °C drop per cycle in first 20 cycles. Annealing temperature is maintained at 55°C through the last 10 cycles.), and 30 s at 72 °C, and an additional 2 min at 72 °C using tag enzyme (NEB Inc.). If EAP was identified, subsequent DNA library construction would be performed; otherwise, WGA and direct mutation detection would be performed on single sperm or PB2 to select GAP. The WGA products of identified GAPs would then be used for subsequent DNA library construction.
DNA library, NGS, and aneuploidy detection
The MALBAC product was used for DNA library construction by NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, Inc.). The whole genome of each sample was sequenced on the Illumina HiSeq X Ten platform (USA) at a 2× sequencing depth. The reads sequenced after filtering were mapped to the human reference genome hg38. Embryo aneuploidy was detected using the method reported by Hou et al. [19]. After normalization, the raw coverage depth was calculated, followed by division of the average sequencing depth of the samples, as well as the corresponding normalization factor. With this method, deletions and duplications not smaller than 10 Mb and a mosaic rate higher than 30% could be detected (the detection limit).
Haplotype analyses based on SNP linkage
Using the NGS results, we analyzed the SNP markers around the mutation sites. In this study, only SNPs with more than three covered reads were included in our analyses. Proband embryos and germline cells carrying the pathogenic mutations (EAP and GAP) allowed us to identify the mutant and the wild-type alleles. The data obtained from other embryos were compared with the identified mutant and wild-type alleles to construct haplotypes using scHaplotyper software [20]. SNP haplotypes within 3 Mb upstream and 2 Mb downstream of PKD1 were illustrated by scHaplotyper, and SNP markers within 1 Mb of PKD1 were selected for further analysis. Through these analyses, the mutation carrier status of embryos can be determined. To overcome the errors introduced by allele dropout (ADO), we used the hidden Morkov model (HMM) to correct the embryo haplotypes [20].
Results
Mutation site detection and proband identification
Due to single-cell WGA allele dropout, SNP linkage analyses are essential for embryo diagnosis. For families with de novo mutations, identifying a proband for haplotype analyses is necessary. Specific primers were designed and touchdown PCR was performed to detect the mutation site directly and to identify the “proband.” To avoid pseudogenes interference, multiple primers were designed and the amplicon contains at least one base to distinguish PKD1 gene from pseudogenes. Touchdown PCR is another way to guarantee specific amplification. As seen in Fig. 1a, direct detection for case 7 on PKD1, c.914G>A shows that all embryos except for E7-2 were affected. To figure out whether the genetic status of E7-2 was interfered by allele dropout, E7-1 was taken as the “proband” (EAP) for further haplotype analyses. Unlike case 7, some cases (Fig. 1b and c) did not have EAP. At this point, to avoid allele dropout, we can resort to backup SAP (Fig. 1b, Sperm32) or PAP (Fig. 1c, E2-1 PB2) to complete haplotype analyses. Other cases used the same strategy to identify the “proband.” Case 1 and case 8 used SAP while cases 5, 6, and 9 used EAP to accomplish subsequent haplotype analyses (Table 2).

Direct detection of PKD1 mutation by Sanger sequencing following specific touchdown PCR. a PKD1 mutation detection of case 7. b PKD1 mutation detection of case 3. c PKD1 mutation detection of cycle 1 of case 2. Red arrows indicate the mutations. Red dotted lines represent mutation sites while green dotted lines represent the adjacent sites that can distinguish PKD1 from the pseudogenes
PGT-M stimulation cycles and aneuploidy detection
In Fig. 2a, a total of 13 PGT-M stimulation cycles were performed; 156 oocytes were obtained. The fertilization rate was 69.23% (108 fertilized oocytes) and the blastocyst formation rate was 31.48%. All 34 embryos were biopsied at the blastocyst stage for subsequent genetic diagnosis. The biopsied blastocysts had an embryo aneuploidy rate of 26.47%. At a 2× sequencing depth, 20 M reads pair could be obtained from each sample and an accurate aneuploidy rate would be calculated. Here, we elucidate CNV analyses using case 9 as an example. As shown in Fig. 2b, chromosome aneuploidy analyses were performed for all 12 embryos of case 9. Ten embryos were without chromosomal abnormalities, while E9-4 and E9-10 were considered not suitable for embryo transfer due to aneuploidy.

Mutation diagnosis based on direct detection and haplotype analyses
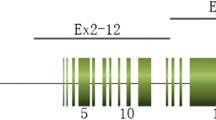
Direct mutation detection was performed for all 34 embryos and detection success rate was 100%. To avoid allele dropout, haplotype analyses were necessary for verification. Schematic haplotype analyses based on gametes carrying the mutant allele or affected embryos were shown in Fig. 3a and b respectively. Case 9 was taken as an example to present the haplotype analysis results based on NGS data (Fig. 3c and d). E9-2 was first identified as proband through specific touchdown PCR, and then SNP markers within 3Mb upstream and 2Mb downstream of PKD1 mutation were analyzed by scHaplotyper software (Fig. 3c). Based on the genotype of E9-2 SNP markers, the haplotype carrying the mutation could be deduced. As shown in Fig. 3c, the haplotypes of the embryos were compared to the EAP (E9-2) to ascertain whether the embryo carries the PKD1 mutation. For further analysis, SNP markers within 1 Mb of PKD1 mutation were selected to confirm the carrier status of embryos (Fig. 3d). For example, the genotype of rs73485872 was C/T for paternal, C/C for maternal, and C/T for E9-2, so we could deduce that the base of T inherited from the father linked with the mutation. Embryos whose genotypes were C/T inherited the base of T from the father carrying the mutant allele, while embryos with C/C genotypes inherited the base of C from the father carrying the wild-type allele. With this approach, all the genotypes of SNPs of the embryos inherited from the father were deduced.

Schematic haplotype analyses and case 9 results from NGS data. a Schematic haplotype analyses using single sperms or PB2 as proband. b Schematic haplotype analyses based on affected embryos. c Haplotype analysis results of case 9 using E9-2 as the proband (EAP). This image is a visualization of the haplotype data within a range of 3 Mb upstream or 2 Mb downstream from the PKD1 gene using scHaplotyper. Red represents the mutant haplotype deduced from EAP. Green represents the wild-type haplotype. d Representative SNP linkage analyses for case 9. Five upstream and five downstream SNP markers in the adjacent regions of the PKD1 gene (1 kb upstream/downstream regions) were selected to identify the disease-carrying allele in each embryo. “W” represents wild type and red cross represents the mutation. RSID, reference SNP cluster ID; CHROM, chromosome number; POS, genomic position; M, maternal; P, paternal; NA, not available
To overcome the errors introduced by allele dropout, we used the hidden Morkov model (HMM) to correct the embryo haplotypes [20]. Through the above analyses, E9-4, E9-8, and E9-10 inherited the mutant allele, and E9-1, E9-3, E9-5, E9-6, E9-7 E9-9, and E9-12 inherited the wild-type allele. These were consistent with the mutation detection results (Table 2). The haplotype analyses of E9-11, however, were inconsistent with its mutation detection result. We suspected that WGA nonuniformity led to allele dropout in direct mutation detection of the embryos.
Overall, after comparing the results of haplotype analyses to direct mutation detection for all diagnosed embryos (Table 2), two embryos were identified with ADO during mutation detection and the ADO rate was 5.9% (2/34). It suggested that haplotype analyses are necessary for accurate diagnosis.
Clinical results
From the 13 IVF-ICSI cycles, seventeen mutation-free and euploid confirmed embryos from 8 couples could be transferred and the embryo transplantable rate was 50% (17/34) (Table 2). Four couples (cases 2, 3, 7, 9) received frozen embryo transfer (FET) and were clinically pregnant. One couple (case 6) was planning for transfer, while 3 couples (cases 1, 5, 8) suffered implantation failure (Table 2). Unfortunately, case 3 had an early miscarriage and the SNP array study of the fetus tissue did not show chromosomal abnormality. The amniocentesis was performed on case 2 and case 7 at 20 weeks of gestation, and the PGT-M results were reconfirmed by PKD1 gene mutation detection of gDNA from the cultured amnion cells (Fig. 4a and b). Of note, no mutation was observed from the cultured amnion cells gDNA. Two healthy neonates in case 2 and case 7 were born successfully.

Mutation detection by Sanger sequencing following specific touchdown PCR of the cultured amnion cells. a Amnion cell mutation (PKD1, c.10453_10454insAGCCC) detection of case 2. b Amnion cells mutation (PKD1, c.914G>A) detection of case 2
Discussion
ADPKD is a single-gene autosomal dominant kidney disease characterized by renal cysts and progression to renal failure. PGT-M is an effective strategy to prevent its transmission from one generation to another. However, it was reported that ADPKD shows a high de novo mutation rate as high as 10–25% [21], which poses a challenge to linkage analyses of PGT-M. To overcome this difficulty, we developed a comprehensive and wide-applicable strategy for these PKD1 patients. The workflow of this strategy, using gamete or embryo as proband for linkage analyses (GEPLA), is shown in Fig. 5. First, single sperms of male patients and PB2 of female patients were collected as backups respectively after ICSI and fertilization. Second, the affected embryo or gamete carrying mutant allele as proband is identified. All blastocysts were biopsied and the whole genome of the samples was amplified, and then the carrier status of each embryo could be determined through Sanger sequencing upon specific touchdown PCR. Once an embryo carrying the mutant allele is identified, it could be regarded as EAP for following haplotype analyses. If no EAP was identified in a family, a supplemental procedure would be performed. Mutation detection for the collected sperms or PB2 would be conducted to determine the “proband.” Third, DNA library was constructed and the NGS at a 2× depth was performed to sequence the blastocyst WGA product. Lastly, the aneuploid status of each embryo was determined based on NGS data. To avoid allele dropout, SNP-based haplotype analyses were performed. According to EAP or GAP with known carrying status of the mutation, linkage analyses were carried out to identify the parental haplotype and the carrier status of the embryos was deduced consequently. In addition to the direct mutation site detection, the diagnosis of embryos was double-checked with SNP-based haplotype analysis.

Flow chart of the flexible strategy. The solid line shows the preferential strategy and the dotted line shows the supplemental strategy
Because of the pseudogenes, mutation detection of PKD1 gene is very challenging [22]. Our mutation detection PCR was performed using specific primers and touchdown procedure. Specific primers were designed against diverse sequences and the amplicon contained at least one specific site to distinguish PKD1 from its pseudogenes. As shown in Fig. 1a and b, Sanger sequencing showed the pseudogenes were excluded. In case 2, the c.10453_10454insAGCCC mutation of PKD1 gene was in the terminal of the duplicated region without pseudogenes interference [23]. We detected all mutations successfully and only 2 embryos had allele dropout. Our preferential selection of EAP was a success and this novel mutation detection method is both convenient and cost-effective. If no EAP can be used, gametes carrying the mutant allele can be chosen for haplotype analyses. Our double-checking strategy containing mutation detection and haplotype analyses is applicable to almost all de novo PKD1 mutations and can ensure accuracy for genetic diagnosis results.
In summary, our method, GEPLA, provides a comprehensive and specific strategy for preimplantation genetic diagnosis of patients with de novo PKD1 mutations or lacking positive family member. Furthermore, this strategy offers new insights into the preimplantation genetic testing of other single-gene disorders with de novo mutations.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
References
Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329(5):332–42.
Gabow PA, Chapman AB, Johnson AM, Tangel DJ, Duley IT, Kaehny WD, et al. Renal structure and hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1990;38(6):1177–80.
Torres VE, Erickson SB, Smith LH, Wilson DM, Hattery RR, Segura JW. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988;11(4):318–25.
Gabow PA, Duley I, Johnson AM. Clinical profiles of gross hematuria in autosomal dominant polycystic kidney-disease. Am J Kidney Dis. 1992;20(2):140–3.
Murphy G, Tzamaloukas AH, Listrom MB, Gibel LJ, Meleg Smith S, Gardner JKD. Nephrotic syndrome and rapid renal failure in autosomal dominant polycystic kidney disease. Am J Nephrol. 1990;10(1):69–72.
Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151–60.
Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell. 1995;81(2):289–298.
Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–42.
Zimmerman K, Yoder BK. SnapShot: sensing and signaling by cilia. Cell. 2015;161(3):692–692.e1.
Bogdanova N, Markoff A, Gerke V, McCluskey M, Horst J, Dworniczak B. Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics. 2001;74(3):333–41.
Eisenberger T, Decker C, Hiersche M, Hamann RC, Decker E, Neuber S, et al. An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS One. 2015;10(2):e0116680.
De Rycke M, Georgiou I, Sermon K, et al. PGD for autosomal dominant polycystic kidney disease type 1. Mol Hum Reprod. 2005;11(1):65–71.
Verlinsky Y, Rechitsky S, Verlinsky O, Ozen S, Beck R, Kuliev A. Preimplantation genetic diagnosis for polycystic kidney disease. Fertil Steril. 2004;82(4):926–9.
Iliuta IA, Kalatharan V, Wang K, Cornec-le Gall E, Conklin J, Pourafkari M, et al. Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017;28(9):2768–76.
Shi H, Niu W, Liu Y, et al. A novel monogenic preimplantation genetic testing strategy for sporadic polycystic kidney caused by de novo PKD1 mutation. Clin Genet. 2020;e13871.
Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205–12.
Wang Y, Zhu X, Yan Z, Zhi X, Guan S, Kuo Y, et al. Novel PGD strategy based on single sperm linkage analysis for carriers of single gene pathogenic variant and chromosome reciprocal translocation. J Assist Reprod Genet. 2020;37(5):1239–50.
Niederberger C, Pellicer A, Cohen J, et al. Forty years of IVF. Fertil Steril. 2018;110(2):185.
Hou Y, Fan W, Yan L, Li R, Lian Y, Huang J, et al. Genome analyses of single human oocytes. Cell. 2013;155(7):1492–506.
Yan ZQ, Zhu XH, Wang YQ, et al. scHaplotyper: haplotype construction and visualization for genetic diagnosis using single cell DNA sequencing data. BMC Bioinformatics. 2020;21(1).
Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67(5):792–810.
Rossetti S, Hopp K, Sikkink RA, Sundsbak JL, Lee YK, Kubly V, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23(5):915–33.
Perrichot R, Mercier B, Quere I, Carre A, Simon P, Whebe B, et al. Novel mutations in the duplicated region of PKD1 gene. Eur J Hum Genet. 2000;8(5):353–9.
Acknowledgements
We thank all the staffs in the Center for Reproductive Medicine of Peking University Third hospital, for supporting all the procedures of IVF-PGT.
Funding
This project is funded by Beijing Municipal Science and Technology Commission (Z191100006619073, Z191100006619075).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuqian Wang and Fan Zhai are joint First Authors and contributed equally to this work.
Supplementary information
Supplemental Fig. 1
Pedigree charts for the nine cases. Filled symbols represent patient affected with ADPKD; open symbols represent wild-type individuals with respect to ADPKD. Circles and squares indicate females and males, respectively. Couples indicated by asterisks asked for PGT-M treatment. (PNG 208 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Zhai, F., Guan, S. et al. A comprehensive PGT-M strategy for ADPKD patients with de novo PKD1 mutations using affected embryo or gametes as proband. J Assist Reprod Genet 38, 2425–2434 (2021). https://doi.org/10.1007/s10815-021-02188-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-021-02188-z