
Abstract
Protoplast systems are essential for genome-editing and gene silencing technologies. In brown algae, protoplast isolation has been hampered by protocols that use non-commercial enzymes or crude extracts. This study is the first to report the production of protoplasts from cell-filament suspension cultures of the brown alga, Hecatonema terminale (Kützing) Kylin, using different mixtures of commercial enzymes and chelation pre-treatment. In this study, mixture A (cellulase RS and alginate lyase) with chelation pre-treatment produced the highest number of protoplasts (3.52 ± 0.23 × 105 protoplasts g−1 FW). Chelation pre-treatment showed high effects on all kinds of enzyme mixtures. The effects of these different mixtures were examined by two-way ANOVA. We also investigate the optimal protoplast density and regeneration medium for protoplast regeneration. Of 16 combinations for regeneration media, RM1 with lowest initial protoplast density (2.4 × 103 protoplasts mL−1) showed the highest value (74%) of final plating efficiency (FPE) after 13 days of culture. The well-defined heterotrichous thalli with phaeophycean hairs were clearly distinguished after 22 days of culture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protoplasts are naked living plant cells lacking a cell wall; these cells are potentially totipotent and represent an important biological tool for genetic improvement, tissue culture, and physiological studies (Reddy et al. 2008; Baweja et al. 2009). Their utility in genome-editing and gene silencing technologies has led to a reemergence of protoplast systems over the past few years (Burris et al. 2016). The development of this type of systems is based on the establishment of reproducible protocols for protoplast isolation (Bhojwani and Razdan 1996).
In brown algae, protoplast isolation has been reported in 25 species (Polne-Fuller et al. 1986; Chen and Shyu 1994; Reddy et al. 2008), mainly commercial and anatomically complex species such as Undaria pinnatifida (Xiaoke et al. 2003) and Saccharina japonica (Inoue et al. 2011). In these studies, the complex cell walls were digested using alginate lyases or crude extracts from either marine bacteria or the digestive systems of herbivorous marine invertebrates together with commercial cellulases (Reddy et al. 1994, 2008). However, these alginate lyases are not commercially available, which makes the isolation process expensive and time consuming because they have to be produced. In addition, the activities of the crude extracts fluctuate over time (Cocking 1972; Fitzsimons and Weyers 1985; Kloareg et al. 1989), resulting in low or no reproducibility of the results. Thus, protoplast isolation protocols using commercial enzymes are fundamental for the development of protoplast systems in brown algae.
Hecatonema terminale (Kützing) Kylin is a widespread filamentous and heterotrichous brown seaweed characterized by more or less compact basal discs with radiating branched uniseriate filaments, true phaeophycean hairs, occasionally intercalary longitudinal divisions, and plurilocular sporangia (Womersley 1987). The reasons for choosing this species for this study were (1) there are no previous reports on protoplast isolation; (2) its primitive anatomy which is ideal for protoplast research (Mejjad et al. 1992); and (3) the availability of cell-filament suspension cultures that provide a constant source of tissue and rapidly growing cells (Doelling and Pikaard 1993; Wang et al. 2015).
In this study, we tested and selected commercial lytic enzymes for protoplast production from cell-filament suspension cultures of H. terminale as a first step in the development of an ideal protoplast system in brown algae. In addition, we described the regeneration processes of protoplasts isolated with the best enzymatic mixture.
Materials and methods
Hecatonema terminale was collected by scuba diving at 1 m depth off of Chuja island, Jeju, Korea, on June 26, 2013. Filaments of H. terminale were cultured in 100 × 40 mm Petri dishes containing PES medium (Provasoli 1968) under a 14:10-h light/dark photoperiod with a light intensity of 40 μmol photons m−2 s−1 at 20 °C. The medium was renewed every 3 weeks. After 3–4 months in culture, plants were accumulated and transferred into 500-mL flat-bottomed round flasks filled with PES medium under aeration with a light intensity of 40–72 μmol photons m−2 s−1 under the same temperature and photoperiod. The air was sterilized using 0.22-μm surfactant-free cellulose acetate (SFCA) syringe filters (Corning, Germany). One month later, plants were finally transferred to 1-L flat-bottomed round flasks and cultured under the same conditions. The medium was renewed every 2 weeks. Clone spheres were broken up monthly using an Ultra–Turrax homogenizer (T25, Ika–Works Inc., USA) in order to maintain homogenous cell-filament suspension cultures.
Identification of the culture strain
Cultures maintained in 60 mm × 15 mm Petri dishes without agitation were used for morphological characterization. Photomicrographs were taken using a Leica inverted microscope (DMi8; Leica, Germany) equipped with a Leica DFC450C camera. Taxonomic identification was performed according to Clayton (1974) and Womersley (1987) and confirmed molecularly. Genomic DNA extraction, PCR amplification, DNA purification, and sequencing were performed according to Bustamante et al. (2016) using cultured samples. The plastid rbcL and mitochondrial COI genes were amplified using the primer combinations described by Kogame et al. (1999) and Lane et al. (2007). The amplified gene sequences were compared to the GenBank nucleotide database using the BLAST program (Altschul et al. 1997).
Protoplast isolation and purification
The commercially available cell wall lytic enzymes used for this study included cellulase Onozuka RS and R-10, macerozyme R-10 (Yakult Co. Ltd., Japan), and alginate lyase (Sigma-Aldrich, USA). Different enzyme combinations were evaluated and the optimal enzyme mix was selected for the highest protoplast yield (Table 1). Various concentrations for each enzyme within optimal mixture were also tested.
Protoplast isolation was performed by the protocols of Benet et al. (1997) and Coelho et al. (2012). Approximately 100–300 mg plants from 1-L round flasks were incubated in a 0.22-μm filter–sterilized enzymatic solution (400 mM NaCl, 130 mM MgCl2·6H2O, 22 mM MgSO4, 160 mM KCl, 2 mM CaCl2, and 10 mM MES; pH 6.3) containing different combinations of enzymes at 15 °C with shaking at 70 rpm for 15 h in the dark. Protoplasts were filtered by using a 25-μm nylon mesh to remove undigested filaments and concentrated by centrifugation at 100×g for 10 min. Protoplast yields were estimated by using a hemocytometer (Marienfeld, Germany) with an Olympus microscope (BX51TRF; Olympus, Japan) and expressed as protoplasts g−1 fresh weight (FW). Average protoplast size was calculated by using ImageJ 1.46r software (Abràmoff et al. 2004) based on 100 cell measurements for each repetition. Also, we tested the effect of chelation pre-treatment for each enzyme mixture. Chelation pre-treatment was conducted with a calcium-chelating solution [665 mM NaCl, 30 mM MgCl2·6H2O, 30 mM MgSO4, 20 mM KCl, and 20 mM ethylene glycol–bis(β–amino–ethyl ether)–N,N,N′,N′–tetraacetic acid tetrasodium salt (EGTA–Na4) as the calcium chelator; pH 5.5] for 20 min prior to enzymatic digestion (Coelho et al. 2012).
Protoplasts were washed twice with enzymatic solution and laid on a 0.9 M sucrose solution. After centrifugation for 10 min at 100×g, the purified protoplasts appeared as a brown band between the sucrose and enzymatic solution phases. They were harvested and washed once with enzymatic solution.
Viability and cell wall removal/formation
The viability of protoplasts was assessed by the exclusion of 0.05% Evans Blue and red chlorophyll autofluorescence using a Leica DMi8 inverted microscope fitted with a Leica EL6000 external light source for fluorescence excitation and equipped with a 470/40-nm emission filter and a 515-nm suppression filter.
The removal (true protoplast) and formation (regeneration) of cell wall were confirmed by staining the cells with 0.01% calcofluor white M2R (Sigma-Aldrich, USA) and examining them using a Leica DMi8 inverted microscope equipped with a 360/40-nm emission filter and a 425-nm suppression filter. A bursting assay was also carried out according to Björk et al. (1990), to verify the absence of cell walls.
Protoplasts regeneration experiments
Protoplasts were dispensed into 1 mL of regeneration medium in 24-well tissue culture test plates. To investigate the optimal protoplast density and regeneration medium for protoplast regeneration, the 16 combinations from four initial protoplast densities (2.4 × 103, 9 × 103, 7 × 104, and 1 × 105 protoplasts mL−1) and four regeneration media were tested at 20 °C in the dark (Table 2). After 2 days in the dark, osmotic pressure was reduced slowly using PES medium and cultures were gradually exposed to a final light intensity of 40 μmol photons m−2 s−1 14:10-h light/dark photoperiod at 20 °C. The medium was renewed every week. The response of cultured protoplasts was assessed using a modified definition of the term “final plating efficiency” (FPE, Ochatt and Power 1992) that is based on distinct developmental stages post first mitotic division. In this study, FPE was defined as the percentage of the originally plated protoplast (Po) that had proliferated into uniseriate filaments with at least one branch (Pfb), which is the basic anatomic architecture of this species and occurs, for H. terminale, at least after 13 days of culture. FPE (%) was calculated using the following equation:
Statistical analysis
Normality and homoscedasticity were examined by using the Shapiro–Wilk and Levene tests, respectively, prior to conducting two-way analysis of variance (ANOVA) for the comparison of protoplast yield under different enzyme mixtures and chelation pre-treatment. One-way ANOVA was performed to examine the effects of different enzyme concentrations in the optimal mixture. Tukey’s post hoc test was used when the results were significant. Effect sizes (Sullivan and Feinn 2012) were presented for ANOVA analysis as ω2. The significance threshold was set at p = .01 in order to reduce the true Type I error rate (at least 7%, but typically close to 15%) (Sellke et al. 2001). Protoplast isolation was repeated three times in each treatment. All statistical tests were performed by using Minitab 17.1 (State College, PA, USA).
Results
Strain identification
The vegetative characteristics of Hecatonema terminale are shown in Fig. 1. Cultures (with or without aeration) did not present reproductive structures during the study. Suspension cultures consisted of small clumps approximately 1 mm in diameter and branched free-living filaments. Our morphological identification of H. terminale was confirmed molecularly. In molecular analyses, a 1245-bp portion of the 1476-bp rbcL gene was sequenced for the strain (MH500017) of H. terminale. The rbcL sequence of our strain was 99% identical to Hecatonema sp. 86 (AF207802.1). Our COI–5P gene sequence (MH500016) was 99% identical to all H. terminale strains reported by Peters et al. (2015).

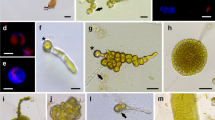
Thallus of Hecatonema terminale. a and b Cultures without aeration. c Thallus with phaeophycean hair (arrow). d Filament with longitudinal divisions (arrows). e A 2-week-old suspension culture in 1-L flat-bottomed round flasks with aeration. The scale in a is 1 cm; the scale in b is 1 mm; the scale in c is 200 μm; the scale in d is 50 μm
Protoplast isolation using enzymes
Our various mixtures of four enzymes (cellulase RS, cellulase R-10, macerozyme R-10, and alginate lyase) showed that Mixture A (cellulase RS and alginate lyase) with chelation pre-treatment produced the highest number of protoplasts (3.52 ± 0.23 × 105 protoplasts g−1 FW), followed by Mixture D (cellulase RS, cellulase R-10, and alginate lyase) with chelation pre-treatment (2.75 ± 0.15 × 105 protoplasts g−1 FW), Mixture E (cellulase RS, macerozyme R-10, and alginate lyase) with chelation pre-treatment (1.20 ± 0.06 × 105 protoplasts g−1 FW), and Mixture A (cellulase RS and alginate lyase) without chelation pre-treatment (1.03 ± 0.05 × 105 protoplasts g−1 FW) (Table 4).
Chelation pre-treatment showed high effects on all kinds of enzyme mixtures (Tables 3 and 4). Addition of a calcium-chelating solution prior to enzymatic digestion significantly increased protoplast amount in mixtures A and D by 2.4- and 2.0-fold, respectively. Interestingly, chelation pre-treatment resulted in lower release of protoplasts from enzyme mixtures that did not contain alginate lyase (Table 4).
Cellulose degradation started to occur from 3 h after treatment of enzyme mixtures (Fig. 2a–c). Although some cell walls were not degraded completely, protoplasts were released through apical or one–sided holes in the cell wall (Fig. 3b). Protoplasts were spherical shape with several discoid chloroplasts (Fig. 3a, c). They were 11.6 ± 2.5 μm in diameter. True protoplast percentages were 98–100% with calcofluor white staining and the bursting assay (Figs. 2c and 3c), while spheroplasts (cells with partially removed cell walls) were 2% (Fig. 3d). The viability of freshly isolated protoplasts was 99–100% with Evans Blue staining and approximately 98% with red chlorophyll autofluorescence.

Protoplast isolation from Hecatonema terminale after 3 h of incubation. a Thallus with cell wall (blue fluorescence) prior to enzymatic digestion. b Thallus with cellulose degradation after 3 h of enzymatic digestion. c True protoplasts (spherical cells with red autofluorescence) released from thalli. The scales in a, b, and c are 200 μm

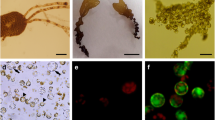
Protoplast isolation from Hecatonema terminale. a Freshly isolated protoplasts. b Protoplast release. c True protoplasts (red autofluorescence) and a spheroplast (arrow). d Closer view of a spheroplast. P protoplast, CWG cell wall ghost. The scale in a is 100 μm; the scale in b is 40 μm; the scale in c is 50 μm; the scale in d is 20 μm
Protoplast cell wall formation
After 3 h of culture in 10 μg mL−1calcofluor white, cell wall formation started with a blue fluorescence spot (positive staining) in one pole of the cell (Fig. 4a–c). After 72 h, additional blue fluorescence spots were detected on 90% protoplasts. Blue fluorescence spots spread across the protoplast surface and covered the whole cell (Fig. 4d–f). After 96 h of culture, 81% of protoplasts with positive staining were regenerated by their cell wall formation.

Cell wall formation of protoplasts from Hecatonema terminale cultured in RM1 at 2.4 × 103 protoplasts mL−1. a Light microscope image of a freshly isolated protoplast. b Fluorescence image of protoplast at initial stage. c Fluorescence image of cell wall formation after 3 h of culture. d Fluorescence image of cell wall formation after 6 h of culture. e Fluorescence image of cell wall formation after 12–48 h of culture. f Fluorescence image of cell wall formation after 72 h of culture. Areas showing bright blue fluorescence indicate cellulose deposition. The red autofluorescence of the chlorophyll reveals areas without cell wall. The scale in a is 20 μm; the scales in b, c, d, e and f are 10 μm
Protoplast regeneration
After cell wall formation, protoplasts underwent cell division in all combinations of initial protoplast densities and regeneration media. After 13 days of culture, protoplasts in RM1 with lowest initial protoplast density (2.4 × 103 protoplasts mL−1) showed the highest value (74%) of FPE. However, protoplasts in RM3 were poorly developed by the formation of short unbranched filaments (Fig. 5).

Final platting efficiency (FPE) of protoplasts from Hecatonema terminale cultured at four initial protoplast densities (2.4 × 103, 9 × 103, 7 × 104, and 1 × 105 protoplasts mL−1) and in RM1 (circles), RM2 (squares), and RM4 (triangles). RM3 was excluded because of poor protoplast development
Protoplast produced a bud in one pole of the cell prior to first asymmetric cell division (Fig. 6a, b). After 13 days of culture, buds developed into prostrate uniseriate filaments with one (11%) or more (54%) primary branches (Fig. 6c, d). After 17 days of culture, secondary and tertiary branches were produced. The well-defined heterotrichous thalli with phaeophycean hairs were clearly distinguished after 22 days of culture (Fig. 6e, f). Mikrosyphar-like plants also developed from protoplasts but in low percentage (9%) (Fig. 6g).

Regeneration stages of protoplasts from Hecatonema terminale. a Bud in one pole of the cell prior to first cell division. b First asymmetric cell division. The arrow indicates the division plane. c 3-celled stage. d Branched filament after 13 days of culture. e A phaeophycean hair (arrow) arising from the initial protoplast (asterisk) in a regenerated plant. f Whole plant regeneration after 22 days of culture. gMikrosyphar-like plant developing from a protoplast at 17 days of culture. The scales in a, b, and c are 10 μm. The scale in d is 60 μm. The scale in e is 20 μm. The scale in f is 400 μm. The scale in g is 100 μm
Discussion
The cell walls of brown algae are comprised mainly of alginate and fucoidans and a small amount (1–8%) of cellulose. Although they do not produce xylans, they synthesize fuco–glucurono–xylans, which have been proposed to cross-link cellulose fibers and alginate gels (Cronshaw et al. 1958; Kloareg and Quatrano 1988). Filamentous brown algae tend to have simple cell wall compositions as low or no presence of sulfated fucans in Ectocarpales; (e.g., Kloareg and Quatrano 1988) and the use of simple enzyme combinations on them suggests low alginate content (Chen and Shyu 1994). In this study, the highest protoplast yield for Hecatonema terminale was obtained using a simple mix of commercial cellulase RS (1%) and alginate lyase (3 U mL–1). Although we are using the mixtures of commercial enzymes, our protoplast yield from H. terminale is superior to the amount of protoplasts reported for Sphacelaria sp. (Ducreux and Kloareg 1988) and in the range of values obtained for Pylaiella littoralis (Mejjad et al. 1992). These differences could be due to interspecific variation of cell wall composition and the type of enzyme mixtures used.
Cellulases RS and R–10 are the most common commercial enzymes used for isolating protoplasts from brown algae. In our study, cellulase RS was more effective than cellulase R-10, showing a 9-fold increase in protoplast yield. Removing cellulase RS from the best enzyme mixture (mixture A) yielded no protoplasts (data not shown). The xylanase and cellulase activity (measured as filter-decomposing-activity) in the RS preparation is 5- and 2-fold higher than in the cellulase R–10 preparation, respectively (Thayer 1985). The main structural role of cellulose and the presence of fuco–glucurono–xylans in the cell wall of brown algae explain the effectiveness of cellulase RS in H. terminale protoplast production. The addition of macerozyme R–10 to the enzyme mixture containing cellulase RS and alginate lyase did not improve protoplast yield. The effect of macerozyme R–10 inclusion in enzyme formulations has not been previously studied in brown algae. Reddy et al. (2006) found that this enzyme was inappropriate for protoplast isolation from Ulva and Monostroma. They demonstrated that macerozyme R–10 is unnecessary in enzyme mixtures if the algal cell walls do not contain pectin or its derivatives, which is the case for brown algae. Our results also suggest that macerozyme R–10 can be excluded when isolating protoplasts from Phaeophyceae.
The incomplete cell wall digestion reported in this study has been described in Sphacelaria sp. (Ducreux and Kloareg 1988), Pylaiella littoralis (Mejjad et al. 1992), and female gametophyte of Macrocystis pyrifera (Varvarigos et al. 2004). However, this was not an impediment to obtain true and viable protoplasts from H. terminale. Considering the fast degradation of cellulose during the isolation process, incomplete digestion might be due to the specificity of the commercial alginate lyase used in this study. According to the manufacturer, this lyase is a mannuronate lyase, which exhibits inefficient alginate gel disruption in comparison to the high activity of guluronate lyases (Formo et al. 2014). Despite this limitation, our results indicate that alginate lyase from Sigma, in combination with cellulase RS, is effective in protoplast isolation from H. terminale.
The addition of cation chelators has been reported to have a positive effect on protoplast production in Ectocarpales (Mejjad et al. 1992; Coelho et al. 2012) and Laminariales (Butler et al. 1989; Kloareg et al. 1989). However, this positive effect might be also affected by the concentration of the chelator, pH of the solution, incubation time, and alginate content in the sample (Butler et al. 1989; Chen and Shyu 1994). In our study, the effect of chelation pre-treatment was dependent on the specific type of enzyme mixture. Only combinations containing alginate lyase showed significant increases following incubation in the chelating solution.
Regeneration ability is one of important parts in protoplast systems (Bhojwani and Razdan 1996). Protoplast was capable of cell wall regeneration and division, although cell division was affected by the initial protoplast densities and regeneration media. In filamentous brown algae, although single initial protoplast density in range from 1 × 102 to 5 × 105 protoplasts mL−1 has been used, its effect never has been tested (Ducreux and Kloareg 1988; Mejjad et al. 1992; Kuhlenkamp and Müller 1994; Benet et al. 1997). Our results showed an optimum density of 2.4 × 103 protoplasts mL−1 for protoplast regeneration. Higher initial protoplast densities decrease the regeneration ability probably because fast depletion of nutrients (Davey et al. 2005) or toxins secreted by cells undergoing necrosis (Yeong et al. 2008). Our initial protoplast densities were also tested with different regeneration media (Table 2). RM1 medium containing CaCl2 was the most effective for protoplast regeneration. Calcium may be an important factor for protoplast regeneration of H. terminale. Calcium is known as a crucial regulator in plant growth and development (Hepler 2005). RM4 medium without enrichment produced the lowest FPE. This suggests that enrichment might be necessary for increasing protoplast regeneration.
Cell wall regeneration started after 3 h of culture, which is similar to what was reported for female gametophyte of Macrocystis pyrifera (Varvarigos et al. 2004). Complete cell wall regeneration was delayed 1 or 2 days compared with Sphacelaria sp., Pylaiella littoralis, and M. pyrifera (Ducreux and Kloareg 1988; Mejjad et al. 1992; Varvarigos et al. 2004). However, regeneration time for whole plant was similar to Sphacelaria sp. (Ducreux and Kloareg 1988) and faster than ones in Laminaria digitata and M. pyrifera (Benet et al. 1997). The regeneration pathway of H. terminale was mainly unipolar and characterized by an asymmetric first cell division after budding and outside of the protoplast. This is distinguished from the protoplast development reported for Sphacelaria sp., Ectocarpus siliculosus, L. digitata, and M. pyrifera (Ducreux and Kloareg 1988; Mejjad et al. 1992; Kuhlenkamp and Müller 1994; Benet et al. 1997; Varvarigos et al. 2004). The occurrence of Mikrosyphar-like plants has been only reported for Hecatonema streblonematoides (Loiseaux 1970). Further studies may be necessary to link this stage to the life cycle of H. terminale.
In conclusion, although a previous study using commercial enzymes reported low viability and survival of protoplasts from brown algae (Chen and Shyu 1994), our results show that true protoplasts with high viability and regeneration capacity can be obtained by a simple mixture of commercial enzymes (cellulase RS and alginate lyase) with chelation pre-treatment.
References
Abràmoff MD, Magalhães PJ, Ram SJ (2004) Image processing with ImageJ. Biophoton Int 11:36–42
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Baweja P, Sahoo D, García-Jimenez P, Robaina RR (2009) Seaweed tissue culture as applied to biotechnology: problems, achievements and prospects. Phycol Res 57:45–58
Benet H, Ar Gall E, Asensi A, Kloareg B (1997) Protoplast regeneration from gametophytes and sporophytes of some species in the order Laminariales (Phaeophyceae). Protoplasma 199:39–48
Bhojwani SS, Razdan MK (1996) Plant tissue culture: theory and practice, a revised edition. Elsevier, Amsterdam
Björk M, Ekman P, Wallin A, Pedersén M (1990) Effects of growth rate and other factors on protoplast yield from four species of Gracilaria (Rhodophyta). Bot Mar 33:433–439
Burris KP, Dlugosz EM, Collins AG, Stewart CN, Lenaghan SC (2016) Development of a rapid, low-cost protoplast transfection system for switchgrass (Panicum virgatum L.). Plant Cell Rep 35:693–704
Bustamante DE, Won BY, Cho TO (2016) The conspecificity of Pterosiphonia spinifera and P. arenosa (Rhodomelaceae, Ceramiales) inferred from morphological and molecular analyses. Algae 31:105–115
Butler DM, Ostgaard K, Boyen C, Evans LV, Jensen A, Kloareg B (1989) Isolation conditions for high yields of protoplasts from Laminaria saccharina and L. digitata (Phaeophyceae). J Exp Bot 40:1237–1246
Chen CS, Shyu JF (1994) Isolation of protoplast from four species of brown algae. Bot Bull Acad Sinica 35:95–104
Clayton MN (1974) Studies on the development, life history and taxonomy of the Ectocarpales (Phaeophyta) in southern Australia. Aust J Bot 22:743–813
Cocking EC (1972) Plant cell protoplasts – isolation and development. Annu Rev Plant Physiol 23:29–50
Coelho SM, Scornet D, Rousvoal S, Peters N, Dartevelle L, Peters AF, Cock JM (2012) Isolation and regeneration of protoplast from Ectocarpus. Cold Spring Harb Protoc 2012:361–364
Cronshaw J, Myers A, Preston RD (1958) A chemical and physical investigation of the cell walls of some marine algae. Biochim Biophys Acta 27:89–103
Davey MR, Anthony P, Power JB, Lowe KC (2005) Plant protoplasts: status and biotechnological perspectives. Biotechnol Adv 23:131–171
Doelling JH, Pikaard CS (1993) Transient expression in Arabidopsis thaliana protoplasts derived from rapidly established cell suspension cultures. Plant Cell Rep 12:241–244
Ducreux G, Kloareg B (1988) Plant regeneration from protoplast of Sphacelaria (Phaeophyceae). Planta 174:25–29
Fitzsimons PJ, Weyers JDB (1985) Properties of some enzymes used for protoplast isolation. In: Pilet PE (ed) The physiological properties of plant protoplasts. Springer, Berlin, pp 12–24
Formo K, Aarstad OA, Skjåk-Bræk G, Strand BL (2014) Lyase-catalyzed degradation of alginate in the gelled state: effect of gelling ions and lyase specificity. Carbohydr Polym 110:100–106
Hepler PK (2005) Calcium: a central regulator of plant growth and development. Plant Cell 17:2142–2155
Inoue A, Mashino C, Kodama T, Ojima T (2011) Protoplast preparation from Laminaria japonica with recombinant alginate lyase and cellulase. Mar Biotechnol 13:256–263
Kloareg B, Quatrano RS (1988) Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Oceanogr Mar Biol Annu Rev 26:259–315
Kloareg B, Polne-Fuller M, Gibor A (1989) Mass production and regeneration of protoplasts from Macrocystis pyrifera. Plant Sci 62:105–112
Kogame K, Horiguchi T, Masuda M (1999) Phylogeny of the order Scytosiphonales (Phaeophyceae) based on DNA sequences of rbcL, partial rbcS, and partial LSU nrDNA. Phycologia 38:496–502
Kuhlenkamp R, Müller DG (1994) Isolation and regeneration of protoplast from healthy and virus-infected gametophytes of Ectocarpus siliculosus (Phaeophyceae). Bot Mar 37:525–530
Lane EC, Lindstrom SC, Saunders GW (2007) A molecular assessment of northeast Pacific Alaria species (Laminariales, Phaeophyceae) with reference to the utility of DNA barcoding. Mol Phylogenet Evol 44:634–648
Loiseaux S (1970) Notes on several Myrionemataceae from California using culture studies. J Phycol 6:248–260
Mejjad M, Loiseaux-de-Goër S, Ducreux G (1992) Protoplast isolation, development, and regeneration in different strains of Pilayella littoralis (L.) Kjellm. (Phaeophyceae). Protoplasma 169:42–48
Ochatt SJ, Power JB (1992) Plant regeneration from cultured protoplasts of higher plants. In: Murray MY, Fowler MW, Warren GS (eds) Plant biotechnology: comprehensive biotechnology (2nd Suppl.). Pergamon Press, New York, pp 99–127
Peters AF, Couceiro L, Tsiamis K, Küpper FC, Valero M (2015) Barcoding of cryptic stages of marine brown algae isolated from incubated substratum reveals high diversity in Acinetosporaceae (Ectocarpales, Phaeophyceae). Cryptogam Algol 36:3–29
Polne-Fuller M, Saga N, Gibor A (1986) Algal cell, callus, and tissue cultures and selection of algal strains. Nova Hedwigia 83:30–36
Provasoli L (1968) Media and prospects for the cultivation of marine algae. In: Watanabe A, Hattori A (eds) Cultures and collections of algae. Proceedings of the U.S.–Japan Conference, Hakone, pp 63–75
Reddy CRK, Fujita Y, Bajaj YPS (1994) Somatic hybridization in algae. In: Bajaj YPS (ed) Biotechnology in agriculture and forestry, somatic hybridization in crop improvement I, vol 27. Springer, Berlin, pp 483–502
Reddy CRK, Dipakkore S, Kumar GR, Jha B, Cheney DP, Fujita Y (2006) An improved enzyme preparation for rapid mass production of protoplasts as seed stock for aquaculture of macrophytic marine green algae. Aquaculture 260:290–297
Reddy CRK, Gupta MK, Mantri VA, Bhavanath J (2008) Seaweed protoplast: status, biotechnological perspectives and needs. J Appl Phycol 20:619–632
Sellke T, Bayarri MJ, Berger JO (2001) Calibration of p values for testing precise null hypotheses. Am Stat 55:62–71
Sullivan GM, Feinn R (2012) Using effect size-or why the P value is not enough. J Grad Med Educ 4:279–282
Thayer SS (1985) Protoplasts for compartmentation studies. In: Linskens HF, Jackson JF (eds) Cell components. Springer, Berlin, pp 54–65
Varvarigos V, Katsaros C, Galatis B (2004) Radial F-actin configurations are involved in polarization during protoplast germination and thallus branching of Macrocystis pyrifera. Phycologia 43:693–702
Wang H, Wang W, Zhan J, Huang W, Xu H (2015) An efficient PEG-mediated transient gene expression system in grape protoplasts and its application in subcellular localization studies of flavonoids biosynthesis enzymes. Sci Hortic 191:82–89
Womersley HBS (1987) The marine benthic flora of southern Australia. Part II. South Australian Government Printing Division, Adelaide
Xiaoke H, Xiaolu J, Huashi G (2003) Isolation of protoplast from Undaria pinnatifida by alginate lyase digestion. J Ocean Univ Qingdao 2:58–61
Yeong HY, Khalid N, Phang SW (2008) Protoplast isolation and regeneration from Gracilaria changii (Gracilariales, Rhodophyta). J Appl Phycol 20:641–651
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01058359); the National Institute of Biological Resources (NIBR) funded by the Ministry of Environment (MOE) of Korea (NIBR No. 2015-01-204); and by Marine Biotechnology Program grants (20170431 and PJT200669) funded by the Ministry of Oceans and Fisheries of Korea to Tae Oh Cho.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Avila-Peltroche, J., Won, B.Y. & Cho, T.O. Protoplast isolation and regeneration from Hecatonema terminale (Ectocarpales, Phaeophyceae) using a simple mixture of commercial enzymes. J Appl Phycol 31, 1873–1881 (2019). https://doi.org/10.1007/s10811-018-1660-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-018-1660-6