
Abstract
Betaphycus Doty, Eucheuma J. Agardh, and Kappaphycus Doty (Solieriaceae, Gigartinales) are the three most commercially important seaweed genera that produce carrageenan. In the present study we provide mitogenomes of Betaphycus gelatinus, Eucheuma denticulatum and Kappaphycus alvarezii. The mitogenomes of these three species contain a set of 50 genes, including 24 protein-coding genes, 2 rRNA genes, and 24 tRNA genes. The mitogenome length ranges from 25,198 bp (Kappaphycus alvarezii) to 25,327 bp (Eucheuma denticulatum). As compared with the previous published mitogenomes of Florideophyceae species, only the species in Gelidiaceae and Pterocladiaceae have smaller mitochondrial genome size than these reported here. At the junction of two transcription units, we identified a stem-loop structure in six representative Gigartinales species, which is presumed to play an important role in the replication and transcription of mitochondrial genes. In Gigartinales the difference in gene order among the four Solieriaceae (B. gelatinus, E. denticulatum, K. alvarezii, K. striatus) and other two Gigartinales species (Chondrus crispus and Mastocarpus papillatus) can be explained by inversion of two tRNA genes. Collinearity analysis of the 12 mitochondrial genomes of Florideophyceae showed considerable sequence synteny across all the species compared, with the exception of a highly variable region between atp6 and rpl20 genes. Phylogenetic analyses based on 21 shared mitochondrial genes showed that the four Solieriaceae species form one clade (Solieriaceae clade). Within this clade, B. gelatinae is basal relative to the other three species. The genus Kappaphycus is more closely related to Eucheuma than Betaphycus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The family Solieriaceae (Gigartinales: Florideophyceae) consists of approximately 19 genera, mostly distributed in the warm-temperate and tropical waters throughout the world (http://www.algaebase.org/). This family of red algae provide important raw materials for carrageenan production (Bixler 1996), which is widely used in food and chemical industries. In Solieriaceae, Betaphycus Doty, Eucheuma J. Agardh, and Kappaphycus Doty are the three most commercially important genera that predominately produce beta-carrageenan, iota-carrageenan, and kappa-carrageenan (Pereira et al. 2015; Tirtawijaya et al. 2016). These three species are the main cultivars in Southeast Asia and China. Many studies on these species focus on the improvement of farming and culture techniques. However, genomic studies on these species are relatively limited.
Species included in these three genera show high phenotypic plasticity and lack of diagnostic morphological features; therefore, all three groups are similarly taxonomically challenging (Doty and Norris 1985). In addition, the plasticity often results in the cultivation of mixed populations by local farmers (Tan et al. 2013), which hinders the processing of the breeding. In recent years, DNA sequences from nucleus, plastid, and mitochondrion have been utilized for phylogenetic research and taxonomic study that are changing the traditional understanding. The application of cox1, cox2–3 spacer datasets, or rbc L has provided insight into the taxonomy of Kappaphycus, Eucheuma, and Betaphycus (Fredericq et al. 1999; Zuccarello et al. 2007; Tan et al. 2013; Dumilag et al. 2014; Lim et al. 2017). However, the above researches were all just based on limited sequence data, which might not exhaust the phylogenetic relationship and taxonomic study of Kappaphycus, Eucheuma, and Betaphycus, and further comprehensive probe is still urgently needed.
Mitochondrial genes are considered to be a valuable alternative to nuclear genes, for the origin of mitochondria being probably concomitant with that of eukaryotic cells (Martin and Müller 1998; Vellai et al. 1998); thus, they facilitate the investigation of the molecular evolutionary relationship. As compared with the small fragments containing genes, the complete mitochondrial genome data contains more phylogenetic signals, and it can provide more precise phylogenetic results than short fragments. Moreover, the mitochondrial DNA generally characterized by uniparental inheritance, limited recombination, and compact genome structure (Bonen 1998). Therefore, mitochondrial DNA is considered an efficient molecular marker for phylogenetic studies (Boore and Brown 1998; Burger et al. 2003).
Recently, owing to decreasing costs of high-throughput sequencing, the next-generation sequencing (NGS) and third-generation sequencing technologies have facilitated the efficient production of genome-scale data (Heather and Chain 2016). Analysis of the genome-scale data could provide information on gene content, structure, and organization, and provide an opportunity of resolving complex phylogenetic relationships (Zhang et al. 2012; Ma et al. 2014). Several recent surveys have used mitochondrial genome reconstructions and phylogenetic relationships of red algae species, and summarized various aspects of mitochondrial genome structure (Yang et al. 2015; Boo et al. 2016; Ng et al. 2017).
In the present study, we first determined the complete mitochondrial genome sequences of three species of Solieriaceae (Betaphycus gelatinus, Eucheuma denticulatum, and Kappaphycus alvarezii). Thereafter, we obtained information about gene content, structure, and organization of these mitochondrion genomes, and compared them with previously published mitochondrial genomes of Florideophyceae species. Further, we used the novel mitochondrial genome data for the reconstruction of phylogenetic relationships and taxonomic study about red algae species with emphasis on relationship of Solieriaceae species.
Materials and methods
Sample collection and DNA extraction
One individual from each species (Betaphycus gelatinus, Eucheuma denticulatum, and Kappaphycus alvarezii), collected from Hainan Province (Table S1), was used to sequence the mitogenome. Genomic DNA was extracted from approximately 1 g of frozen tissue using a modified cetyltrimethylammonium bromide (CTAB) method (Sun et al. 2011). DNA quality and quantity were determined by the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA).
High-throughput sequencing, assembly, and mitochondrial genome annotation
Three short-insert libraries were constructed from approximately 5 μg of purified DNA for each species. Paired-end reads were extracted from Illumina’s HiSeq × Ten system (Illumina, USA). Approximately 9 Gb of paired-end (125 bp) sequence data was randomly extracted from the total sequencing output, as input into NOVOPlasty (Dierckxsens et al. 2017) to assemble the mitochondrial genome. Kappaphycus striatus (GenBank accession KF833365) was used as the seed sequence. The protein-coding genes and ribosomal RNA of the three species were annotated from K. striatus using Geneious R10 (Biomatters Ltd., New Zealand; available from http://www.geneious.com/). To predict tRNA genes, the mitochondrial genome sequences were submitted to the tRNAscan-SE ver. 1.21 (http://lowelab.ucsc.edu/tRNAscan-SE/) (Schattner et al. 2005). The physical map of the mitogenomes was prepared for visualization using Organellar Genome DRAW (OGDRAW) (Lohse et al. 2007). Secondary DNA structures were predicted by using RNAfold webserver (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi).
Comparative genome analysis
Comparisons of the mitogenome sequences with previously published Florideophyceae mitogenome sequences were generated using progressive Mauve genome aligner version 2.4.037 (Darling et al. 2004) at the default settings.
Estimation of substitution rates
To test the selection pressures and gene evolution rates of the four Solieriaceae species (B. gelatinus, K. striatus, K. alvarezii, and E. denticulatum), ratios of nonsynonymous (dN) and synonymous (dS) substitutions were analyzed using PAML v4.4 package (Yang 2007), with Chondrus crispus as the reference. The program yn00 was employed to estimate dN, dS, and dN/dS under the F3 × 4 substitution matrix using the Nei-Gojobori method. Kruskal-Wallis tests were conducted using the R software package (http://www.r-project.org).
Phylogenetic analysis
Phylogenetic analyses were conducted based on the following dataset: 21 conserved protein-coding genes (atp4, atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rpl16, rps3, rps12, sdh2, sdh3, sdh4) from 41 red algal mitochondrial genomes available at the NCBI GenBank (including three species included in the present study) (Table S2). Three protein-coding genes (including rpl20, rps11, and tatC) were not used to construct the phylogenetic tree because they were pseudogenes or lost from the mitogenomes of some red algae (Table S2). The protein sequences were aligned using MEGA 5.0 software (Tamura et al. 2011) and subsequently edited and trimmed manually using BioEdit version 7.0.5.3 (Hall 1999). The concatenated alignments were generated and poorly aligned regions were removed using the Gblocks server (http://phylogeny.lirmm.fr/phylo_cgi/one_task.cgi?task_type=gblocks) (Castresana 2000), and the alignment was reduced from 5913 to 4246 position. The best-fit model for maximum likelihood (ML) was selected using ProtTest 3.4.2 (Darriba et al. 2011) based on Akaike Information Criterion (AIC). The phylogeny was inferred by the ML search and ML bootstrap analysis using RAxML (Stamatakis 2006). Bootstrap probability values were run with 1000 replicates under the CpREV + G + I + F model. The Bayesian inference (BI) was performed using MrBayes v. 3. 1.2 (Huelsenbeck and Ronquist 2001). The phylogenetic analysis was performed using two independent runs with four Markov Chains Monte-Carlo for 1000,000 generations. Output trees were sampled every 100 generations. The phylogenetic analysis was run until the average standard deviation of split frequencies was below 0.01 and the first 25% of samples was removed as burn-in. The protein sequences of Cyanidioschyzon merolae (NC_000887) was used as outgroup.
Results and discussion
Genome composition and structure
The mitogenomes of B. gelatinae (MF680514), E. denticulatum (MF680515), and K. alvarezii (NC_031814) were assembled as a single circular molecule using NGS methodologies. Genes are encoded on both the heavy and light strands, with approximately the same encoding proportion on each strand in two opposite but major transcriptional directions (Fig. S1–S3). The composition and structure of the four representative species in Solieriaceae are summarized in Table 1. All four Solieriaceae mitogenomes contain a set of 50 genes, including 24 protein-coding genes, two rRNA genes, and 24 tRNA genes. One intron is inserted in the tRNA-Ile gene (Table 1; Fig. S1–S3). The number of genes in B. gelatinae, K. alvarezii, K. striatus, and E. denticulatum is the same, but different from that of C. crispus and Mastocarpus papillatus (Table 1; Fig. S1–S3).
The mitogenome length ranges from 25,198 bp (K. alvarezii) to 25,327 bp (E. denticulatum) (Table 1). As compared with the C. crispus (family Gigartinaceae), Gracilaria changii (Gracilariaceae) and M. papillatus (family Phyllophoraceae), both from the order Gigartinales, the four Solieriaceae species show smaller mitochondrial genome size (Table 1). Compared with the previously published mitochondrial genome of Florideophyceae species, the mitochondrial genome length of Solieriaceae are relatively smaller. Only one species each in Gelidiaceae and Pterocladiaceae show smaller mitochondrial genome sizes than that of the ones reported in the present study (Table S4). The overall GC content ranges from 29.70% (B. gelatinae) to 30.3% (E. denticulatum), which is comparable to the other Florideophyceae species (Table S4). In addition, the mitogenome of K. striatus is 44 bp longer than that of K. alvarezii, mainly because of differences in length of the intergenic regions. The intergenic regions of K. alvarezii is 4.96% whereas that of K. striatus 5.13% (Table 1).
Protein-coding gene
All protein-coding genes use the mold genetic code, which differs from the universal code only in the use of TGA as tryptophan (Trp). The protein-coding gene lengths of all 24 protein-coding genes in K. striatus and K. alvarezii are identical (Table 2). Significant difference in gene length was observed among B. gelatinae, E. denticulatum, and the two Kappaphycus species. Fifteen of the 24 protein-coding genes differ in length among B. gelatinae, E. denticulatum, and the two Kappaphycus species (Table 2).
The start codon for all mitochondrial genes in E. denticulatum is ATG. The start codon for nearly all mitochondrial genes in B. gelatinae, K. alvarezii, and K. striatus is ATG, the exception being tatC, which uses the TTG as the start codon. There are two stop codons, the most predominant of which is TAA, with nad3, atp4, cox1, nad4L, tatC, and rpl20 using TAG.
As compared with C. crispus (family Gigartinaceae) from the same order Gigartinales, the gene content of the protein-coding gene of the four Solieriaceae species are almost the same as that of C. crispus (Table 1). Besides the 24 protein-coding genes mentioned above, the mitochondrial genome of M. papillatus (family Phyllophoraceae) possesses one tatA gene, which is not observed in the other Gigartinales species. All protein-coding genes had the same ATG start codon in G. changii mitochondrial genes. The start codon for nearly all mitochondrial genes in C. crispus and M. papillatus is ATG, whereas that of the tatC gene is GTT in C. crispus and ATC in M. papillatus. The most predominant stop codon is TAA, whereas sdh3 and sdh4 uses TAG in C. crispus, and nad4, rps11, sdh3, and rpl20 uses TAG in M. papillatus.
Secondary structure
Consistent with previously published mitochondrial genomes of Gigartinales, such as C. crispus (Leblanc et al. 1995), K. striatus (Tablizo and Lluisma 2014), and M. papillatus (Sissini et al. 2016), the mitochondrial genome of B. gelatinae, E. denticulatum, and K. alvarezii consists of two obvious transcriptional units oriented in opposite transcriptional directions (Fig. S1–S3). At the demarcation points of these two transcriptional units, located within the intergenic regions of the mitogenome, we identified a long stable stem-loop (trnA-trnS) (Fig. 1). These structures are complete inverted-repeat sequences containing polymers of A and T (Fig. 1). This secondary structure is similar to the displacement-loop (D-loop), which is known to be a control region in the mitochondrial genome of mammals (Clayton 1982; Taanman 1999), wherein it is located at the junction of two transcriptional units and is presumed to activate the initiation of DNA replication. Moreover, it contains the major promoter for transcription. In addition, a short hairpin structure (cob-trnL) can be found in the opposite stem-loop.

A comparison of the sequences of the a stem-loop and b hairpin structures from six Gigartinales mitochondrial genomes
Among all the sequenced mitochondrial genomes of brown and green algae, no similar sequences have been identified. Thus, it appears that such stem-loop structures occur only in algal species of the phylum Rhodophyta. At present, mitochondrial sequences from nearly 80 red algae species have been published. Similar stem-loop and hairpin structures have been identified in the mitochondrial genomes of in orders Ceramiales, Gelidiales, Gracilariales, Palmariales, Halymeniales, Plocamiales, Rhodymeniales, Batrachospermales, and Bangiales (Figs. S4 and S5). As indicated by Zhang et al. (2012), such stem-loop structures can lead to the deletion or base mutation of DNA fragments during PCR, thus creating gaps in the whole-genome sequence. Therefore, homologous amplification and long-PCR strategies have been widely used in previous attempts to obtain complete mitochondrial DNA sequences. This might be a possible reason for the incomplete nature of the sequences obtained. However, high-throughput sequencing can effectively avoid this problem. Due to its high sequence coverage, this approach can correct random mismatches during the PCR process (caused by the instability of Taq DNA polymerase). In addition, it can facilitate the complete sequencing of fragments containing complicated secondary structures. Moreover, the sequence determined in the present study will provide an important source of data for future research on the mitochondrial genome of Solieriaceae.
Gene order
In Gigartinales the difference in gene order between the four Solieriaceae and the other two Gigartinales species (C. crispus and M. papillatus) in this region can be explained by the inversion of two tRNA genes: trnY (gua) and trnR (ucu) (Fig. 2). Detailed characterization of the gene order based on synteny analyses for 12 mitochondrial genomes of Florideophyceae showed strong collinearity among species in most of regions, with all 12 genomes showing similar gene numbers and genomic organizations (Fig. S6). A highly variable region was identified between the atp6 and rpl20 genes (Fig. S6). Moreover, it was not surprising that this region is located at the junction of two transcription units. Within this variable region, there are considerable differences in the type, number, and order of tRNA genes among the different species (Fig. S6). In general, the mitochondrial genomes of Florideophyceae species are highly conserved and there is considerable sequence synteny across all the species. The region near the junction of the two transcriptional units is the only highly variable region in the mitochondrial genomes.

Comparison of six Gigartinales mitochondrial genomes using Mauve software
Gene substitution rates
With the inclusion of the results of the present study, the sequences of only four mitochondrial genomes in the family Solieriaceae have been published to date. We calculated and compared the gene substitution rates of all the 24 protein-coding genes in the four species based on nucleotide sequences. The dN/dS values differed significantly (P < 0.001) among each gene (Fig. 3). The dN/dS values of all the protein-coding genes in the four species were far below one, suggesting the existence of purify selection on these genes. The atp9 genes have the lowest substitution rates (0.0034), and the same situation is observed in previous studies on Gelidiales species (Boo et al. 2016). Generally, if a gene has a low substitution rate, this indicates that the gene is important or the gene-related function is more conserved. Our results confirmed the amino acid sequence stability of the atp9, and the gene product indicates that this gene is highly conserved.

Ratio of nonsynonymous (dN) versus synonymous substitutions (dS) value for 24 protein-coding genes in four Solieriaceae mitochondrial genomes. A: Betaphycus gelatinus; B: Eucheuma denticulatum; C: Kappaphycus striatus; D: Kappaphycus alvarezii
Within the ATP synthase group, atp8 (0.3897) had higher mean dN/dS ratios than atp4 (0.3248), atp6 (0.0695), and atp9 (0.0093), suggesting that there has been differential selection, and that each gene evolved independently within the functional group.
Additionally, cytochrome oxidase genes (cox1) show lower substitution rates than that of the other genes. This explains why cox1 is widely used as an ideal tool for phylogenetic analysis (Conklin et al. 2009). Cox2, cox3, nad1, nad2, and nad3 show similar dN/dS ratios as that of cox1, which indicated that they show a more stable rate of variation at both the nucleotide and amino acid levels. They may thus be suitable markers for detecting molecular evolution and phylogenetic structure in red algae.
Phylogenetic analysis
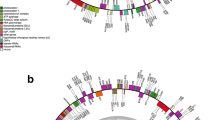
Figure 4 presents the maximum likelihood (ML) and Bayesian inference (BI) tree of 43 Rhodophyta species based on 21 shared mitochondrial protein-coding genes. The topological structure of the phylogenetic trees inferred by ML and BI methods are concordant, with the exception of the branch with low bootstrap support in the Gigartinales clade (see below). The taxa are clearly separated into two clades representing Bangiophyceae and Florideophyceae with high bootstrap and posterior probability values. The Bangiophyceae clade contains 10 species, among which six Pyropia species are divided into two small clades. The Florideophyceae clade containing 32 species can be divided into 16 orders. The phylogenetic relationship of Florideophyceae in this study is corresponded well with the one in Yang et al. (2015) and supported the currently accepted relationships of Rhodophyta.

Phylogenetic tree (maximum likelihood and Bayesian inference) of 43 red algae based on 21 mitochondrial protein-coding genes. The support values for each node are calculated from maximum likelihood bootstrap (MLB) and Bayesian posterior probability (BPP). Asterisks after species names indicate newly determined mitochondrial genome. Red block: topology derived from maximum likelihood analysis; green block: topology derived from Bayesian analysis
The most notable finding of the phylogenetic analysis was related to the resolution of the relationship among Gigartinales species. Gigartinales species separate into two clades. Four Solieriaceae species, E. denticulatum, K. alvarezii, K. striatus, and B. gelatinae form one clade (Solieriaceae clade). Within this clade, B. gelatinae is basal relative to other three species. In addition, K. alvarezii forms a cluster with the congeneric K. striatus. Eucheuma denticulatum emerged as the branch closet to Kappaphycus clade. The phylogenetic relationship of Solieriaceae species constructed by mitochondrial data is well supported by morphological characteristics of the Solieriaceae species (Fig. S7). The second clade consists of C. crispus (Gigartinaceae) and M. papillatus (Phyllophoraceae). However, the position of this clade is uncertain; it is clustered with the Solieriaceae clade in the ML tree (bootstrap support = 48), but clustered with Riquetophycus sp. (KJ398161) in the BI tree (posterior probabilities = 0.57). These phylogenetic relationships were not fully resolved because of the low bootstrap and posterior probability values. Further investigations not only on molecular phylogeny of all members within the genus but also on the cytology and molecular biology will be necessary to test and refine the above research.
References
Bixler HJ (1996) Recent developments in manufacturing and marketing carrageenan. Hydrobiologia 326:35–57
Bonen L (1998) Mitochondrial genomes. Adv Genome Biol 5:415–461
Boo GH, Hughey JR, Miller KA, Boo SM (2016) Mitogenomes from type specimens, a genotyping tool for morphologically simple species: ten genomes of agar-producing red algae. Sci Rep 6:35337
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8:668–674
Burger G, Gray MW, Lang BF (2003) Mitochondrial genomes: anything goes. Trends Genet 19:709–716
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Clayton DA (1982) Replication of animal mitochondrial DNA. Cell 28:693–705
Conklin KY, Kurihara A, Sherwood AR (2009) A molecular method for identification of the morphologically plastic invasive algal genera Eucheuma and Kappaphycus (Rhodophyta, Gigartinales) in Hawaii. J Appl Phycol 21:691–699
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14(7):1394–1403
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18
Doty MS, Norris JN (1985) Eucheuma species (Solieriaceae, Rhodophyta) that are major sources of carrageenan. In: Abbott IA (ed) Taxonomy of economic seaweeds. California Sea Grant College Program, La Jolla, pp 47–61
Dumilag RV, Liao LM, Lluisma AO (2014) Phylogeny of Betaphycus (Gigartinales, Rhodophyta) as inferred from COI sequences and morphological observations on B. philippinensis. J Appl Phycol 26:587–595
Fredericq S, Freshwater DW, Hommersand MH (1999) Observations on the phylogenetic systematics and biogeography of the Solieriaceae (Gigartinales, Rhodophyta) inferred from rbcL sequences and morphological evidence. Hydrobiologia 398:25–38
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT; Nucl Acids Symp Ser 41:95–98
Heather JM, Chain B (2016) The sequence of sequencers: the history of sequencing DNA. Genomics 107:1–8
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Leblanc C, Boyen C, Richard O, Bonnard G, Grienenberger JM, Kloareg B (1995) Complete sequence of the mitochondrial DNA of the rhodophyte Chondrus crispus (Gigartinales). Gene content and genome organization. J Mol Biol 250:484–495
Lim PE, Yang LE, Tan J, Meggs CA, Brodie J (2017) Advancing the taxonomy of economically important red seaweeds (Rhodophyta). Eur J Phycol 52:438–451
Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52:267–274
Ma PF, Zhang YX, Zeng CX, Guo ZH, Li DZ (2014) Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst Biol 63:933–950
Martin WF, Müller M (1998) The hydrogen hypothesis for the first eukaryote. Nature 392:37–41
Ng PK, Lin SM, Lim PE, Liu LC, Chen CM, Pai TW (2017) Complete chloroplast genome of Gracilaria firma (Gracilariaceae, Rhodophyta), with discussion on the use of chloroplast phylogenomics in the subclass Rhodymeniophycidae. BMC Genomics 18:40
Pereira L, Meireles F, Abreu HT, Ribeiro-Claro PJA (2015) A comparative analysis of carrageenans produced by underutilized versus industrially utilized macroalgae (Gigartinales, Rhodophyta). In: Kim S-K, Chojnacka K (eds) Marine algae extracts: processes, products, and applications. Wiley-VCH, Weinheim, pp 277–294
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:686–689
Sissini MN, Navarrete-Fernández TM, Murray EMC, Freese JM, Gentilhomme AS, Huber SR, Mumford TF, Hughey JR (2016) Mitochondrial and plastid genome analysis of the heteromorphic red alga Mastocarpus papillatus (C. Agardh) Kützing (Phyllophoraceae, Rhodophyta) reveals two characteristic florideophyte organellar genomes. Mitochondrial DNA B 1:676–677
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Sun YY, Luo D, Zhao C, Li W, Liu T (2011) DNA extraction and PCR analysis of five kinds of large seaweed under different preservation conditions. Mol Plant Breed 9:1680–1691
Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123
Tablizo FA, Lluisma AO (2014) The mitochondrial genome of the red alga Kappaphycus striatus (“Green Sacol” variety): complete nucleotide sequence, genome structure and organization, and comparative analysis. Mar Genomics 18:155–161
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tan J, Lim P-E, Phang S-M (2013) Phylogenetic relationship of Kappaphycus Doty and Eucheuma J. Agardh (Solieriaceae, Rhodophyta) in Malaysia. J Appl Phycol 25:13–29
Tirtawijaya G, Mohibbullah M, Meinita MDN, Moon IS, Hong Y-K (2016) The ethanol extract of the rhodophyte Kappaphycus alvarezii promotes neurite outgrowth in hippocampal neurons. J Appl Phycol 28:2515–2522
Vellai T, Takacs K, Vida G (1998) A new aspect to the origin and evolution of eukaryotes. J Mol Evol 46:499–507
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang EC, Kim KM, Kim SY, Lee J, Boo GH, Lee J-H, Nelson WA, Yi G, Schmidt WE, Fredericq S, Boo SM, Bhattacharya D, Yoon HS (2015) Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol Evol 7:2394–2406
Zhang L, Wang X, Qian H, Chi S, Liu C, Lui T (2012) Complete sequences of the mitochondrial DNA of the wild Gracilariopsis lemaneiformis and two mutagenic cultivated breeds (Gracilariaceae, Rhodophyta). PLoS One 7(6):e40241
Zuccarello GC, Critchley AT, Smith J, Sieber V, Lhonneur GB, West JA (2007) Systematics and genetic variation in commercial Kappaphycus and Eucheuma (Solieriaceae, Rhodophyta). In: Anderson R, Brodie J, Onsøyen E, Critchley AT (eds) Proceedings of the Eighteenth International Seaweed Symposium. Springer, Dordrecht, pp 417–425
Funding
This work was supported by the China-ASEAN Maritime Cooperation Fund, by Public Science and Technology Research Funds Projects of Ocean (Grant No. 201405020), and by the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, Y., Liu, N., Wang, X. et al. Comparative genomics and systematics of Betaphycus, Eucheuma, and Kappaphycus (Solieriaceae: Rhodophyta) based on mitochondrial genome. J Appl Phycol 30, 3435–3443 (2018). https://doi.org/10.1007/s10811-018-1450-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-018-1450-1