
Abstract
Viscosity is a thermophysical property of paramount importance, being essential for many scientific and industrial applications. The most common instruments for its measurement are glass capillary viscometers. Therefore, the use of capillary viscometers is widespread both in industry and in research. The range of viscosities of interest range from lower than that of water to several orders of magnitude higher values, the measurement of which requires different capillary viscometers. Most of the practical applications concern routine instruments, mainly for quality control. One main issue for the utilization of capillary viscometers relates to the need for their calibration, assuring its traceability to the water primary viscosity standard, to certify its worldwide validity. The present paper focuses on capillary instruments dedicated to perform viscosity measurements on Newtonian organic liquids at atmospheric pressure, as it is assumed that is the most widespread type of application for these viscometers. Capillary viscometry has a completely well-defined working equation, namely, the Hagen–Poiseuille equation. However, the practical performance of the measuring instruments deviates from that working equation. Most of those deviations are currently considered by many users. However, some of those deviations have not reached that status yet, like those concerning the effects due to the surface tension of the sample on the measurements. All these aspects are summarized and analyzed in the present article, together with a brief general description of the most common types of capillary viscometers, namely, the Ostwald and the constant-level or Ubbelohde instruments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Viscosity is recognized to be a crucially important thermophysical property for many engineering and scientific applications. This is especially obvious for what concerns chemical process engineering, both for design areas and in routine applications, like industrial quality control. Capillary viscometry assumes paramount importance as it is the experimental technique generally used to establish the standards to be used both in industry and in scientific research. For those applications, the so-called master or reference capillaries are preferably used by the competent metrological institutions and companies. It is to be noticed that industrial applications of viscosity methods are assuming increasing importance, including for process monitoring. Industrial processes may involve monitoring of reaction processes, quality control, or equipment design. In particular, resort to routine capillaries continues to assume special importance for quality control, for instance, in petrochemical industries. Presently, the above routine measurements are made at atmospheric pressure, which is, therefore, in number, the most important condition for applications of capillary viscometers.
The present paper focuses on the use of capillary viscometers to carry out relative measurements on Newtonian liquids at atmospheric pressure, which the authors consider to be the most important type of application for these instruments.
In the present article, the two most relevant capillary viscometers are described, namely, the Ostwald and Ubbelohde or constant-level instruments, and some of their main issues to be considered when using this technique are reviewed. Gravity force is used to drive the flow of the samples in these types of viscometers and the Hagen Poiseuille equation is the adequate working equation to describe the flow of the liquid samples in these capillaries. The use of other types of capillary viscometers, with different shapes, can make it impractical to use the Hagen Poiseuille equation as they deviate from the ideal model. That equation represents an ideal model, as some of the requirements for its derivation are not met in practice. As a consequence, several deviations to that equation, or additional effects, must be considered. The analysis of those deviations and effects, and how they are usually avoided, calculated, or minimized are an important part of the present article.
It is noticeable that, for each specific capillary viscometer, the calibration, and the quantification of all the mentioned deviations to the basic working equation will play a significant role to ensure the highest possible accuracy of the measurements.
An important issue of capillary viscometry is the unavoidable need to calibrate the instruments, with a mandatory need to guarantee traceability to the water primary standard. In fact, although there are several viscosity reference fluids for a significant range of viscosities, the ultimate worldwide accepted primary standard is water, with a dynamic viscosity value of 1.0016 mPa·s, or a kinematic viscosity of 1.0034 mm2/s at atmospheric pressure. The actual uncertainties of the official values for the viscosity of water, which are currently accepted, are published by the International Organization for Standardization with a value of ± 0.17% [1].
2 Capillary Types
In glass capillary viscometers, the liquids used are generally Newtonian, in which the viscosity is constant at a certain temperature and the viscosity change due to the pressure drop during the measurement may be considered insignificant. Capillary viscometers consist of a reservoir for the liquid sample and a capillary of known dimensions, in particular, its cross section which must also be uniform. The volume of the liquid sample required to operate this type of viscometer is small, and accurate temperature control is necessary. The fluid passes through the capillary tube, and it is necessary to measure the time it takes to pass between the two marks placed at predetermined positions on the top and bottom of the bulb above the capillary tube of the viscometer, useful to indicate the length of the capillary to be used for fluid flow. In automatic systems, the flow time reading is taken by using electronic sensors in fixed positions, such that those marks do not interfere with the detection of the liquid meniscus passage. This makes it possible to measure the flow time much more accurately than before, when it was needed the human intervention to operate a stopwatch. The use of an automatic time measurement system, which must be recalibrated at appropriate time intervals, should have reading resolution not greater than 0.01 s, with a relative measurement uncertainty of no more than 0.02% [2].
These capillary viscometers, made of borosilicate, must be cleaned after handling to preserve the quality of the capillary tube. Even so, whenever they have not been used for some time, they should be cleaned before being used again and care should be taken to keep the calibration constant valid. Cleaning with abrasive systems may alter the characteristics of the glass capillary tube, which will change its calibration constant. The capillary viscometer should be cleaned with a suitable solvent, paying attention to its purity and volatility, and considering the type of fluid used previously in the capillary. If the capillary viscometer is heavily contaminated, a chromosulfuric mixture can be used for about two hours after washing with a solvent. This should be followed by a rinse with distilled or demineralised water, repeated several times. In more extreme cases, hydrochloric acid may also be used [2]. A stream of dry, dust-free air can be used to dry the tube. Use of an oven at a suitable temperature, so as not to damage the glass, or a vacuum desiccator can be a good help for this endeavor.
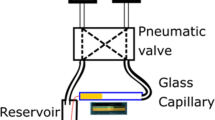
The most used models of capillary viscometers are Ostwald and suspended level instruments. Ostwald viscometers are of the U-tube type (Fig. 1) and the filling procedure is often very difficult, depending on the liquid used [3]. Also, when filling this type of viscometer, it must be placed perfectly aligned vertically since any small inclination may cause an error in the hydrostatic head; also, the volume of liquid to be placed inside this type of capillary viscometer must be exactly known and, when performing measurements at different temperatures from that of the filling, the thermal expansion of the liquid sample should be considered [4]. Previously to measure the falling time of the liquid, it is necessary to apply a pressure, or suction, which causes the liquid to rise above the mark (m1, in Fig. 1) placed at the top of the bulb above the capillary tube. Then, the suction is stopped and the liquid begins to flow due to the gravity force, recording the time it takes to pass between the two marks (m1 and m2, in Fig. 1), above and below the bulb at the top of the capillary tube. In addition, the filling of the Ostwald viscometer should ensure that the meniscus of the liquid column is in the upper bulb (A, in Fig. 1), above the capillary tube, and the other meniscus lies in the lower bulb (B, in Fig. 1) at the bottom side of the U-tube, ensuring that the results are reliable [5].

Diagram of an Ostwald viscometer—Reprinted with permission from the American Chemical Society [3]. Copyright {2024} American Chemical Society
Suspended level capillary viscometers (see Fig. 2) arose from the work carried out by Leo Ubbelohde [3], having as a special aim to avoid the surface tension effects of the flowing liquid film at both the top and the bottom of the capillary tube, which may introduce errors in the viscosity measurements. The application of this type of construction makes it possible to balance the effects of surface tension on both sides of the capillary, since the liquid is at atmospheric pressure at both the top and bottom of the column of liquid passing through the capillary tube. This causes the liquid to be suspended at the exit of the capillary tube and not in direct contact with liquid at the bottom of the tube, as in the case of U-tube Ostwald viscometers [3, 6, 7]. Furthermore, it is noteworthy that in the suspended level, or Ubbelohde viscometers, it is not necessary to determine the volume of the liquid sample introduced in the capillary, which is a significant practical advantage when compared to the Ostwald instrument.

Diagram of the viscometer proposed by Ubbelohde—Adapted with permission from the American Chemical Society [3]. Copyright {2024} American Chemical Society
Among the general types of capillary instruments, used for the measurement of viscosity, it is important to differentiate master viscometers from routine viscometers. Master viscometers are used for rigorous relative measurements of the viscosity of liquids. These are typically longer and with larger bulbs than routine capillaries. These differences are aimed to minimize certain uncertainties, like those arising from deviations to the vertical alignment, residual liquid on the walls, and, most importantly, surface tension effects resulting from the use of fluids with different surface tensions, as mentioned by Kawata et al. [7]. It is also noteworthy that the use of long capillaries with long efflux times tend to minimize the kinetic energy corrections for low Reynolds numbers [7].
Routine capillaries are calibrated using reference fluids, whose viscosity has been determined by master capillaries. The present article is particularly dedicated to the use of routine capillaries.
3 Working Equation
The working equation for the utilization of the capillary method to measure the viscosity is based on an ideal model for the description of the flow of a fluid inside a tube. The flow may be generated by the gravity force or by a pressure difference between the ends of the fluid in the tube. That ideal model is the Hagen–Poiseuille equation, which can be written for a volumetric flow rate, Q, in a tube with internal radius, \(a\), in the form [7]:
where \(\eta\) is the (dynamic) viscosity of the fluid sample. \(\Delta P\) is the pressure drop along the tube of length L. Kawata et al. [7] state that, as the result of more rigorous analysis, Eq. 2 has been most commonly used for capillary viscometers.
In Eq. 2, n and m can be named [7] as end correction and kinetic energy correction factors, respectively. The validity of Eq. 2 is subject to a large number of assumptions, namely, a strait and uniform cross section of the capillary tube; the incompressibility and constant density of the fluid; the Newtonian character of the fluid; a negligible variation of the pressure and the temperature of the fluid; laminar and steady flow and no slip of the fluid at the wall of the capillary [7]. It must be added that in many cases, in particular, regarding the use of capillaries for routine works, the second term in Eq. 2 is negligible.
According to Kawata et al. [7], Eq. 2 and its theoretical restrictions, as indicated above, must be applied for absolute capillary viscometers, which are outside the scope of the present work.
Capillary master viscometers [7] and most of the routine capillary viscometers use the small hydrostatic force due to the volume of fluid placed above the capillary entry as the driving force for the fluid to flow through the capillary. Viscometers in which the flow of the liquid through the capillary occurs because of gravity allow the kinematic viscosity, \(\nu\) to be obtained directly [3,4,5]. The kinematic viscosity and the dynamic viscosity of a liquid are related to its density, \(\rho\) by the expression (3).
If the fluid flows in the capillary under gravity, Eq. 1 can be rewritten as Eq. 4 [5, 8]:
where h stands for the average head of the fluid during the time, t, taken by the volume, V, of the fluid to flow through the capillary and g is the gravitational acceleration.
The factors of the flow time, t, in Eq. 4 are constant for each capillary, and therefore, they can be grouped in a constant \(K\).
Equation (5) can be improved by addition of a new constant, \(E\), to account for a so-called correction for the kinetic energy, resulting in Eq. 6.
Equation (6) was proposed by Cannon et al. [9], who have shown that E is a constant in the most important range of Reynolds number for practical capillary viscometer applications. The corrective term \(E\) may be neglected for flow times greater than a minimum [9, 10], often supplied by the manufacturer of the capillaries. Therefore, if the flow time is greater than the mentioned minimum, Eq. 6 can simply be written as the Eq. 5. This subject is discussed in more detail in Sect. 5.
4 Deviations to the Working Equation and Their Corrections
The performance of real instruments deviates from the ideal model describing the flow of a fluid in a capillary tube. Among the most notorious of those deviations are the kinetic energy, transported by the fluid flow, and from surface tension effects during the flow since both issues were not considered in the derivation of the ideal model. Other deviations arise due to the difficulty in assuring the absolute concordance with the assumptions of the model, like a perfect constancy of the cross section along the length of the capillary used in the measurements. Except for the constant-level or Ubbelohde type of viscometers, the difficulty to realize a constant driving head during the measurements is one of such complications.
All these deviations can be minimized by practical arrangements and, in certain cases, may be amenable to quantitative estimation. In particular, master or reference capillaries are primarily devised with the aim to minimize, or eliminate, in practice,several deviations to the ideal model, as, for example, the surface tension effects.
It is of great importance to note that the calibration of each individual capillary will lead to the same general goal, namely, to minimize or practically eliminate some of the otherwise unavoidable deviations to the theoretical model.
4.1 The Surface Tension Effect
If the upper and lower menisci have different average diameters or if the surface tension of the liquid sample is very different from that of the calibrating fluid, a surface tension correction is necessary [10]. Ubbelohde viscometers are designed to compensate the effect of the surface tension in the measuring bulb by means of a suitable curvature of the suspended level, at the bottom of the capillary instrument. However, the complete compensation of the surface tension effects is not possible to achieve both theoretically and experimentally in standard Ubbelohde viscometers [8]. Thus, a special attention to the effects caused by interfacial tension is necessary to obtain accurate experimental results, as reported by several authors, including Bauer and Meerlender [8], Caetano et al. [11] and Diogo et al. [12]. Significant effects of surface tension on capillary measurements may be expected when the surface tension of the calibrant fluids is very different from that of the samples to be analyzed. In many applications, the liquids whose viscosity is to be measured will have a surface tension much lower than that of water. The same kind of issue can be risen by an opposite situation, as it was observed when an enormous interest in ionic liquids was raised in the scientific community. In fact, ionic liquids often have a very high surface tension, when compared to the commercial standard reference oils frequently used for the calibration of the capillaries [12]. A direct means to assess these surface tension effects may be obtained by performing simultaneous measurements by a method inherently free of surface tension effects, like the vibrating wire technique [13], and by routine capillary viscometers, which has been done in some studies [12,13,14]. A quantitative analysis was carried out by Caetano et al. [11] and by Diogo et al. [12], on the basis of the work developed by Bauer and Meerlender [8]. According to the latter, the surface tension effects on the raw viscosity measurements performed with a suspended level or Ubbelohde capillary can be assessed by including a correction factor χ to the Hagen–Poiseuille equation, such that [8]:
where \({\nu }_{corr}\) is the corrected kinematic viscosity of the test fluid i, \(K\) is the viscometer constant, t is the flow time for fluid i, σ is its surface tension, and ρ is its density. The χ factor is a constant for a given capillary, and it can be obtained by using two viscosity standards (0 and A) according to [11, 12]:
Considering Eq. 4, the correction due to the surface tension effect can be obtained using Eq. 9.
In Eq. 9, the subscript (0) corresponds to the fluid used to calibrate the viscometer, i.e., to determine the constant, \(K\).
4.2 The Kinetic Energy Correction
The kinetic energy correction is one of the most significant corrections applied to the capillary viscosity measurements. In fact, when using this type of viscometers, a part of the employed force is converted to kinetic energy carried by the flow of the liquid [4]. However, the Hagen Poiseuille equation, Eq. 1, assumes that the velocity along the axis of the tube is constant and a parabolic distribution of the flow velocity through the cross section is valid. Nevertheless, there are two different chambers required to measure the pressure drop between both ends of the tube and, consequently, the parabolic distribution of the velocity can be assured only some distance downstream from the inlet of the capillary [4, 7]. Moreover, for the fluid to enter the tube, its streamlines must converge, since the diameter of the inlet chamber is much larger than the diameter of the capillary. At the exit of the capillary, the flow coming from the tube creates a jet into the exit chamber. So, both effects contribute for a measured pressure drop, which is different from what is considered in Eq. 2 [7,8,9]. To eliminate this kinetic energy effect, some viscometers have a reservoir connected to both ends of the capillary; however, it has been shown that some correction to this effect is still necessary [4].
The effects related to the dissipation of kinetic energy at the outlet of the capillary, and the need for a part of the hydrostatic pressure to accelerate the liquid as it enters the capillary, increasing the parabolic velocity profile, can be taken into account using a correction ΔtH to the raw experimental value of the flow time t in the form of Eq. 10 [8]:
This correction is known as the kinetic energy correction, and it can be expressed as Eq. 11 [4, 5, 8, 9]:
where m is a dimensionless parameter depending on the Reynolds number, Re, of the flow inside the capillary and the geometry of the capillary ends, which can be determined experimentally. To simplify, for viscometers with a square-cut capillary outlet, it is usually assumed a complete dissipation of kinetic energy and so the value of unity (m = 1) is assigned to m [8]. In the case of trumpet-shaped outlet, it is usual to use m = 0.037·\(\surd Re\), obtained in an early investigation [8, 9]. Bauer and Meerlender [8] state that, after several viscometer calibrations carried out over many years, it has been found that for Ubbelohde viscometers with these trumpet-shaped ends, the value of m = 0.32 gives satisfactory results (for a Reynolds number lower than 50) [8].
In fact, this correction, ΔtH, is often provided by some suppliers for specific types of viscometers, depending on the corresponding diameter of the capillaries, and on the measured flow time, t, in which case Eq. 10 is rendered unnecessary.
However, it is important to note that although that correction may be written in the form of Eq. 12, B is not a constant, because it depends on m and therefore on the Reynolds number.
Cannon et al. [9] have shown that equating the driving fluid head to the total friction involved in the fluid flow could give rise to another equation, Eq. 6, involving not only the constant \(K\), but also another factor, \(E\), that is constant over a significant range of Reynolds numbers, mostly used in applications. The use of Eq. 6 instead of Eq. 12 has therefore the practical advantage to enable the corrective constant \(E\), for a certain capillary, be obtained by calibration for use over a significant useful range of Reynolds number. Notwithstanding this, the kinetic energy correction term \(E/{t}^{2}\) becomes negligible [10] if the flow time is greater than a certain value.
4.3 The End Effects Correction Related to the Exit of the Capillary
As stated above, end effects are another source of concern regarding the need to assess eventual contributions to the uncertainties for capillary viscosity measurements. During the measurement, the pressure drop of the fluid flow is often measured between two marks corresponding to a chamber in the upper end of the capillary. Consequently, it is crucial to take into consideration the different types of streamlines at the entrance and exit of the capillary for the accurate determination of viscosity, using this type of equipment [4, 7]. The friction caused both in the entry and on the outlet of the capillary has usually been correlated with the kinetic energy of the fluid inside the capillary [7] and is taken into account in the parameter \(E\) in Eq. 6. The parameter \(E\) can be calculated from instrument dimensions, or experimentally. The latter procedure is advised by Cannon et al. [9] as its value is influenced by the form of the capillary ends.
Another way to consider the friction at the ends of the capillary is described by Kawata et al. [7]. It has been devised by Couette (as cited by Kawata et al. [7]), assuming an independent way to describe the end effects on the measurements of viscosity from the outlet of the fluid in the capillary. The main issue seems to depend on the effects on the streamlines at the exit of the capillary, which are a consequence of the specific characteristics of the equipment construction. In fact, an additional pressure drop can occur caused by the change in the flow profile near the center of the chamber. To correct for this, an end correction parameter n is used [7], which depends on the geometry of each capillary [7], and essentially consists in a virtual increase of the capillary length, in order to compensate for the head loss caused by the exit of the fluid from the capillary. Couette was the first author to suggest a virtual increase of the capillary length L, by using the quantity na to quantify these effects (as cited by Kawata et al. [7]). The values of n can vary between 0 and 1.2 [4], and along the years, several authors [15,16,17] have reported several different values for this parameter, obtained either by theoretical or experimental means, as described in [7].
Thus, the Hagen–Poiseuille equation, Eq. 1, can be rewritten considering both kinetic energy and end effects, as presented in Eq. 2 [4, 7] which can also be written in terms of time t, Eq. 13:
4.4 The Buoyancy Correction
The capillary viscosity measurements can also be affected by the buoyancy of the liquid column generated by the ambient air [8]. Although this is a very small effect, that it is often not considered, a correction of the pressure head by a factor \(\left( {1 - {{\rho _{{air}} } \mathord{\left/ {\vphantom {{\rho _{{air}} } \rho }} \right. \kern-\nulldelimiterspace} \rho }} \right),\) where ρair is the density of ambient air and ρ is the density of the liquid, is needed [8]. For most of the applications, the liquid samples have a density ρ close to the standard ρ0 used to calibrate the viscometer [8]. In these cases, the buoyancy correction is usually included in the viscometer constant \(K\), and no correction is applied [8]. On the other hand, when the liquid sample has a density, ρ, considerably different from the reference fluid used to calibrate the viscometer, ρ0, the effect of the air buoyancy must be accounted for, using the Eq. 14 [8].
5 Calibration
The calibration of routine capillary viscometers essentially consists in the determination of the constant \(K\) in Eq. 5, characteristic of each capillary. This procedure is based on the fact that \(K\) is just a constant factor, theoretically related to various parameters appearing in the ideal work equation, some of which would not be easy to independently obtain the corresponding values. In fact, some of the working equation’s inherent prerequisites like the regularity of the inner diameter of a particular capillary is not a simple parameter to obtain experimentally. Therefore, the calculation of the calibration constant in practice enables to obtain a global information on the characteristics of a particular instrument, which otherwise would be very difficult to consider.
The calibration of routine or practical viscometers is usually made using certified reference materials (CRM), available from metrological institutions either state-controlled or private, or by comparison with master (or reference) viscometers, whose constants have often been determined using the step-up procedure [7, 13, 18]. The latter involves the use of several successive capillary viscometers, in some instances, master viscometers, with overlapping ranges of viscosity from the water primary viscosity standard to the high viscosity values targeted [19,20,21] (v.d. Sect. 5.2).
All these different calibration procedures must ensure traceability to the primary reference value for water. In practice, it is necessary to transfer calibration data from the domain of metrological institutions and master capillaries to routine or practical instruments, what will certainly cause some increase to the uncertainty of measurement, presumably not less than ± 1% according to Caetano et al. [20].
Alternatively, the direct calibration of routine capillaries with industrial reference fluids of an appropriate viscosity may involve an uncertainty that, in some cases, may eventually be of a similar order of magnitude as the ones obtainable by the procedures described above, for a considerably lower cost. Additionally, one important characteristic of industrial reference fluids is that they are not proprietary substances, having their properties available in the literature [20].
The calibration constant, \(K,\) can change over time due to modifications on the capillary glass conditions, caused by successive measurements and cleaning procedures, which were mentioned in Sect. 2. For that reason, calibration must be carried out on a regular basis to ensure the maximum quality of the results.
5.1 Primary Standard
The calibration of relative viscometers and viscometry itself is based metrologically on the viscosity of the water value at 20 ºC, under normal atmospheric pressure. This is considered to be the primary standard for viscometry (v.d. Sect. 1), which means that all accredited viscosity measurements must be traceable to this value [1, 8, 19]. The measurements originally published in 1952 by Swindells et al. [22] presented a dynamic viscosity of 1.0019 mPa·s that was corrected due to a 0.012 K gap between the IPTS-48 and the later adopted ITS90 temperature scale at 20 ºC.
The establishment of the reference value for the viscosity of water, and the uncertainty of its measurement have been involved in some discussion along the years, after those results obtained by Swindells et al. were first published [22]. Many measurements of the viscosity of water have been carried out since then. The subject has been discussed in detail by Kenneth Marsh [18]. However, despite the discussion, the original value has remained intact, and is generally assumed as the ultimate reference standard for viscometry, even though its nominal uncertainty has been increased [1, 18]. However, the uncertainty of that value is not very important as long as the measurements of the viscosity of dense fluids are made relative to water, as Kenneth Marsh has pointed out [18].
5.2 Other Standards
In many industrial applications, including process engineering industries, it is frequently needed to measure the viscosity of fluids at certain thermodynamic conditions, often significantly away from those corresponding to the primary standard value. Furthermore, in many instances, those fluids have viscosities very different from that value. This is particularly significant for high viscosity fluids, which are frequently used in industry, requiring routine measurements of viscosities with orders of magnitude several times higher than the primary standard value for water. Therefore, one single reference fluid is not appropriate for calibrating all of the capillary instruments necessary to cover the enormous range of viscosities for fluids in industrial applications with economic interest [20].
Consequently, a step-up procedure is often necessary to certify reference fluids with viscosity near the values required. The step-up procedure is typically carried out using a series of successive master capillaries, with increasing diameters, starting with one adequate to measure the viscosity of water. Each pair of successive capillaries being able to measure the same liquid at the same temperature will therefore enable to transfer each calibration from one instrument to the next. In this, the final uncertainty [18] and the costs of the step-up procedure increase with the number of intermediate master capillaries needed to cover the difference between the water primary standard and the high viscosity target [20]. Therefore, this process implies high costs and lower accuracy results, because of the uncertainty propagation in the procedure, and so, it has been recognized the need for the establishment of reference fluids with higher viscosity, near that of the fluids used in industry at the corresponding practical thermodynamic conditions [19, 21].
The fact that water has a very high interfacial tension, usually much higher than most of the liquids of industrial or scientific interest, poses an additional concern, as the correction for the surface tension effects on capillary measurements is not completely straightforward. This is mainly because these effects must be assessed for each specific capillary, which requires the value of the surface tension of both the calibrant and the test fluid. To circumvent the problem of the high surface tension of water when compared to other fluids, it is essential to use other reference fluids to calibrate the different capillary viscometers needed.
The selection of adequate reference materials must consider important characteristics such as their water solubility, purity, toxicity, availability, low vapor pressure, and importantly, low cost [19]. After the discussion by Künzel et al. in 1987 on reference materials for viscosity, a lot of research work and development contributed to clarify the type of liquids that should be used in calibrating procedures, as well as on quality instrumentation [23].
Several liquids have been proposed as candidates to be reference fluids for viscosity, especially, for high viscosity. These include phthalates [23,24,25,26] and several other kinds of substances, like sebacates [24, 27], benzoates [25], squalane [25, 28, 29], perfluoropolyesters [30], and ionic liquids [31]. It is to be noted that the establishment of reference fluids requires measurements obtained by different experimental methods, which has often been observed. The most relevant and appropriate reference fluids are described below according to their viscosity range.
5.2.1 Low Viscosity Reference Fluids
Along the years, several liquids have been proposed as an alternative to water, such as toluene, alkyl esters, and aliphatic hydrocarbons. Toluene and other hydrocarbons fulfill the requisites to be used as reference materials for low viscosity. Hydrocarbons do not react with most of the materials used in viscometer cells and other ancillary equipment and present a comparatively low surface tension, adequate heat capacity, and thermal conductivity, which are also pointed out as important general requirements for the application as reference materials [23].
Some of these materials are already being used as reference materials for other thermophysical properties [23]. Toluene has been extensively studied through the years extending the applicability of this material as a reference for viscometry [23, 32, 33]. Other hydrocarbons are available with high purity and with good general properties to be a reference fluid, as an alternative to water. Particularly, n-nonane, n-decane, and n-undecane have viscosities very near that of water at room temperature, low water solubility, and a large liquid stability temperature range [23, 34].
5.2.2 High Viscosity Reference Fluids
For moderately higher viscosity than water and toluene at room temperature, higher linear hydrocarbons like n-tetradecane, n-hexadecane, and n-heptadecane could be a good option to be reference materials [14, 35, 36].
For substantially higher viscosities, with several orders of magnitude higher than that of water, some liquids have been proposed to be used as reference fluids with important characteristics to be used as reference materials for high viscosity and at high pressures [11, 13, 19,20,21]. Several studies on Di-isodecylphtalate (DIDP) [11, 19, 20] have contributed to its proposal as a potential high viscosity reference fluid to respond to industrial needs. Subsequently, another compound, namely Tris(2-ethylhexyl) Trimellitate (TOTM), has been indicated as a promising potential standard for higher viscosities at high temperatures and high pressures [13, 21].
It should also be remarked that many of the measurements performed on those liquids have been obtained with techniques that are inherently free of surface tension effects. In addition, measurements with capillaries have also been carried out at atmospheric pressure [12, 13], which is an important contribution to the assessment of eventual surface tension effects on capillary measurements. Naturally, for this purpose, it is necessary to know the corresponding surface tension of the reference liquids, as exemplified by Caetano et al. [11, 12] and discussed in Sect. 5.1.
5.2.3 Certified Calibration Liquids
Notwithstanding the arguments in favor of the pure liquids proposed to be reference fluids for viscosity, their practical use must be validated internationally. Thus, the actual certified reference materials (CRM) manufactured by metrological institutes, or certified companies, with viscosity determined by the mentioned step-up procedures, starting from the primary viscosity standard value, will continue to play an important role to industry for validation needs. These calibration liquids are usually mixtures of oils, with an unknown exact composition [18, 20]. Pure compounds are not usually chosen since it is claimed that their viscosity can be significantly dependent on their purity, in a way that it is not easily predicted [18, 20]. CRMs should be characterized by low affinity to water and high stability over long periods of time [18, 20]. However, these CRMs require attention to the corresponding certificate, provided by the supplier, in particular, to check the expiry date of the certificate values [20, 23]. It is to be noted the need for an extra attention when measuring the viscosity of liquids with a very different surface tension from the CRM used to calibrate the capillaries [12].
6 Summary
Capillary viscometry is an important method for measuring the viscosity of liquids, both in industry and in scientific research. In this work, we describe the most common capillary viscometers, namely, the Ostwald and Ubbelohde or constant-level instruments, and review their main issues to be considered when using this technique. The Hagen Poiseuille equation describes the flow of the liquid samples in these capillaries. However, several deviations to that equation must be considered and their correction and ways to how they are usually avoided, calculated, or minimized are discussed in this article.
Routine capillary instruments need to be calibrated to guarantee traceability to the primary standard as water is the only worldwide accepted primary standard for viscosity.
A step-up calibration procedure is used for calibrating the capillary instruments when the viscosities of the fluid samples are much higher than that of the standard value. This represents a cost increase and time consumption also leading to an extended uncertainty. Therefore, several pure liquids have been proposed to be industrial reference fluids for high viscosity.
Usually, the calibrating liquids have a low surface tension, unlike water, implying the need for the surface tension correction. Other corrections to take into account deviations to the prerequisites of the Clausius–Clapeyron equation, the basis of the theoretical model for the capillary viscometers, like the so-called kinetic energy and the end effects corrections, are also treated in this article.
Capillary viscometers will continue to be used in different sectors, playing an important role to obtain accurate viscosity measurements for industrial purposes, especially, quality control, and for research.
Data Availability
No datasets were generated or analyzed during the current study.
References
Technical Report ISO/TR 3666:1998, Viscosity of Water, International Organization for Standardization, Switzerland.
Determination of kinematic viscosity using the Ubbelohde viscometer. Part I: Apparatus and measurement procedure, DIN 51562–1 (Deutsches Institut für Normung, Berlin, January 1999)
L. Ubbelohde, The principle of the suspended level. Applications to measurement of viscosity and other properties of liquids. Ind. Eng. Chem. 9, 85–90 (1937). https://doi.org/10.1021/ac50106a015
D.S. Viswanath, T.K. Ghosh, D.H.L. Prasad, N.V.K. Dutt, K.Y. Rani, Viscosity of Liquids. Theory, Estimation, Experiment, and Data, 1st edn. (Springer, Berlin, 2007)
F.A. Gonçalves, J. Kestin, J.V. Sengers, Surface-tension effects in suspended-level capillary viscometers. Int. J. Thermophys. 12(6), 1013–1028 (1991). https://doi.org/10.1007/BF00503516
P.A. Young, R.J. Lovell, Introduction to Polymers (CRC Press, Boca Raton, 2011)
M. Kawata, K. Kurase, A. Nagashima, K. Yoshida, Experimental Thermodynamics, The Measurement of Transport Properties of Fluids, vol. III (Blackwell Scientific, Oxford, 1991)
H. Bauer, G. Meerlender, Precise viscosity measurements of Newtonian liquids with v < 1 mm2/s for the selection of suitable standards. Rheol. Acta 23(5), 514–521 (1984)
J.D. Cannon, M.R. Manning, R.E. Bell, The kinetic energy correction and a new viscometer. Anal. Chem. 32(3), 355–358 (1960)
ASTM D446, Standard Specifications and Operating Instructions for Glass Capillary Kinematic Viscometers-IP Designation: 71/2/95 (2012).
F.J.P. Caetano, J.M.N.A. Fareleira, A.C. Fernandes, C.M.B.P. Oliveira, A.P. Serro, I.M. Simões de Almeida, W.A. Wakeham, Diisodecylphthalate (DIDP) a potential standard of moderate viscosity: surface tension measurements and water content effect on viscosity. Fluid Phase Equilib. 245(1), 1–5 (2006). https://doi.org/10.1016/j.fluid.2006.03.012
J.C.F. Diogo, F.J.P. Caetano, J.M.N.A. Fareleira, W.A. Wakeham, Viscosity measurements on ionic liquids: a cautionary tale. Int. J. Thermophys. 35(9), 1615–1635 (2014). https://doi.org/10.1007/s10765-013-1487-y
J.C.F. Diogo, H.M.N.T. Avelino, F.J.P. Caetano, J.M.N.A. Fareleira, W.A. Wakeham, Tris(2-ethylhexyl) trimellitate (TOTM) as a potential industrial reference fluid for viscosity at high temperatures and high pressures: new viscosity, density and surface tension measurements. Fluid Phase Equilib. 418, 192–197 (2016). https://doi.org/10.1016/j.fluid.2016.01.012
T.V.M. Santos, M.F.V. Pereira, H.M.N.T. Avelino, F.J.P. Caetano, J.M.N.A. Fareleira, Viscosity and density measurements on liquid n-tetradecane at moderately high pressures. Fluid Phase Equilib. 453, 46–57 (2017). https://doi.org/10.1016/j.fluid.2017.08.025
J. Kestin, M. Sokolov, W. Wakeham, Theory of capillary viscometers. Appl. Sci. Res. 27, 241–264 (1973)
H. Kawata, M. Kurase, K. Yoshida, K. Utsumi, Flow, Its Measurement and Control in Science and Industry, vol. 1 (Wiley, New York, 1974)
W.N. Bond, Viscosity determination by means of orifices and short tubes. Proc. Phys. Soc. Lond. 34(1), 139–144 (1921). https://doi.org/10.1088/1478-7814/34/1/329
K.N. Marsh, Role of reference materials for the realization of physicochemical properties. Past, present, and future. Pure Appl. Chem. 72(10), 1809–1818 (2000). https://doi.org/10.1351/pac200072101809
F.J.P. Caetano, J.M.N.A. Fareleira, C.M.B.P. Oliveira, W.A. Wakeham, Viscosity of di-isodecylphthalate: a potential standard of moderate viscosity. Int. J. Thermophys. 25(5), 1311–1322 (2004). https://doi.org/10.1007/S10765-004-5740-2
F.J.P. Caetano, J.M.N.A. Fareleira, A.P. Fröba, K.R. Harris, A. Leipertz, C.M.B.P. Oliveira, J.P.M. Trusler, W.A. Wakeham, An industrial reference fluid for moderately high viscosity. J. Chem. Eng. Data 53(9), 2003–2011 (2008). https://doi.org/10.1021/je800059n
W.A. Wakeham, M.J. Assael, H.M.N.T. Avelino, S. Bair, B.A. Bamgbade, H.O. Baled, J. Bazile, F.J.P. Caetano, M.J.P. Comuñas, J. Daridon, J.C.F. Diogo, R.M. Enick, J.M.N.A. Fareleira, J. Fernández, M.C. Oliveira, T.V.M. Santos, C.M. Tsolakidou, In pursuit of a high-temperature, high-pressure, high-viscosity standard: the case of tris(2-ethylhexyl) trimellitate. J. Chem. Eng. Data 62(9), 2884–2895 (2017). https://doi.org/10.1021/acs.jced.7b00170
J.F. Swindells, J.R. Coe, T.B. Godfrey, Absolute viscosity of water at 20-degrees-C. J. Res. Natl. Bur. Stand. 48(1), 1–31 (1952)
C.A.N. De Castro, F.J.V. Santos, J.M.N.A. Fareleira, W.A. Wakeham, Metrology of viscosity: have we learned enough? J. Chem. Eng. Data 54(2), 171–178 (2009). https://doi.org/10.1021/je800528e
S.K. Mylona, M.J. Assael, K.D. Antoniadis, S.K. Polymatidou, L. Karagiannidis, Measurements of the viscosity of bis(2-ethylhexyl) sebacate, squalane, and bis(2-ethylhexyl) phthalate between (283 and 363) K at 0.1 MPa. J. Chem. Eng. Data 58(10), 2805–2808 (2013). https://doi.org/10.1021/je4005245
K.R. Harris, Temperature and pressure dependence of the viscosities of 2-ethylhexyl benzoate, bis(2-ethylhexyl) phthalate, 2,6,10,15,19,23-hexamethyltetracosane (squalane), and diisodecyl phthalate. J. Chem. Eng. Data 54(9), 2729–2738 (2009). https://doi.org/10.1021/je900284z
M.M.A. Motari, M.E. Kandil, K.N. Marsh, Density and viscosity of diisodecyl phthalate C6H4(COOC10H21)2, with nominal viscosity at T = 298 K and p = 0.1 MPa of 87 mPa·s, at temperatures from (298.15 to 423.15) K and pressures up to 70 MPa. J. Chem. Eng. Data 4, 1233–1239 (2007)
X. Paredes, O. Fandiño, A.S. Pensado, M.J.P. Comuñas, J. Fernández, Experimental density and viscosity measurements of di(2ethylhexyl)sebacate at high pressure. J. Chem. Thermodyn. 44(1), 38–43 (2012). https://doi.org/10.1016/j.jct.2011.07.005
M.J.P. Comuñas, X. Paredes, F.M. Gaciño, J. Fernández, J.P. Bazile, C. Boned, J.L. Daridon, G. Galliero, J. Pauly, K.R. Harris, M.J. Assael, S.K. Mylona, Reference correlation of the viscosity of squalane from 273 to 373 K at 0.1 MPa. J. Phys. Chem. Ref. Data 42(3), 6 (2013). https://doi.org/10.1063/1.4812573
M.J.P. Comuñas, X. Paredes, F.M. Gaciño, J. Fernández, J.P. Bazil, C. Boned, J.L. Daridon, G. Galliero, J. Pauly, K.R. Harris, Viscosity measurements for squalane at high pressures to 350 MPa from T = (293.15 to 363.15) K. J. Chem. Thermodyn. 69, 201–208 (2014). https://doi.org/10.1016/j.jct.2013.10.001
H. Baled, D. Tapriyal, B.D. Morreale, Y. Soong, I. Gamwo, V. Krukonis, B.A. Bamgbade, Y. Wu, M.A. McHugh, W.A. Burgess, R.M. Enick, Exploratory characterization of a perfluoropolyether oil as a possible viscosity standard at deepwater production conditions of 533 K and 241 MPa. Int. J. Thermophys. 34(10), 1845–1864 (2013). https://doi.org/10.1007/s10765-013-1500-5
R. D. Chirico, V. Diky, J. W. Magee, M. Frenkel, and K. N. Marsh, Thermodynamic and thermophysical properties of the reference ionic liquid: 1-Hexyl-3-methylimidazolium bis[(Trifluoromethyl)Sulfonyl]amide (Including Mixtures). Part 2. Critical evaluation and recommended property values (IUPAC Technical Report). Pure Appl. Chem. 81(5), 791–828 (2009). https://doi.org/10.1351/PAC-REP-08-09-22
F.J.P. Caetano, J.L. Correia Da Mata, J.M.N.A. Fareleira, C.M.B.P. Oliveira, W.A. Wakeham, Viscosity measurements of liquid toluene at low temperatures using a dual vibrating-wire technique. Int. J. Thermophys. 25(1), 1–11 (2004). https://doi.org/10.1023/B:IJOT.0000022325.10161.39
R.D. Goodwin, Toluene thermophysical properties from 178 to 800 K at pressures to 1000 Bar. J. Phys. Chem. Ref. Data 18(4), 1565–2163 (1989). https://doi.org/10.1063/1.555837
K.N. Marsh, Recommended Reference Materials for the Realization of Physicochemical Properties (Wiley, Oxford, 1987)
T. Klein, S. Yan, J. Cui, J.W. Magee, K. Kroenlein, M.H. Rausch, T.M. Koller, A.P. Fröba, Liquid viscosity and surface tension of n-hexane, n-octane, n-decane, and n-hexadecane up to 573 k by surface light scattering. J. Chem. Eng. Data 64, 4116–4131 (2019)
M.C.M. Sequeira, H.M.N.T. Avelino, F.J.P. Caetano, J.M.N.A. Fareleira, Viscosity and density measurements on liquid n-heptadecane at high pressures. J. Chem. Eng. Data 67, 37–44 (2022). https://doi.org/10.1021/acs.jced.1c00674
Acknowledgements
The authors thank the reviewers of this article for the important comments and suggestions.
Funding
Open access funding provided by FCT|FCCN (b-on). This work was supported by Fundação para a Ciência e a Tecnologia (FCT), Portugal, Projects UIDB/00100/2020, UIDP/00100/2020 and LA/P/0056/2020. M.C.M. Sequeira acknowledges the PhD grant funded by FCT ref. UI/BD/152239/2021.
Author information
Authors and Affiliations
Contributions
All authors contributed actively to the writing of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sequeira, M.C.M., Caetano, F.J.P. & Fareleira, J.M.N.A. Capillary Viscometry for Routine Measurements of Newtonian Liquids. Int J Thermophys 45, 118 (2024). https://doi.org/10.1007/s10765-024-03410-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10765-024-03410-7