
Abstract
Gene expression profiling of lip salivary gland (LSG) has shown that C-X-C motif chemokine 10 (CXCL10) and matrix metalloproteinase 9 (MMP9) expression is upregulated in primary Sjögren’s syndrome (pSS) patients. Although CXCL10 and MMP-9 are both associated with pSS pathogenesis, the potential relationship between these two factors has not been investigated. In this study, we used LSG sections from pSS patients and human salivary gland cell lines to investigate the relationship between CXCL10 and MMP-9. Immunofluorescence analyses revealed that CXCL10 and MMP-9 were co-expressed in the LSG of pSS patients, particularly in expanded ductal cells. Furthermore, RT-qPCR analyses on human salivary gland ductal NS-SV-DC cells confirmed that CXCL10 expression was induced by interferon (IFN)-γ, whereas that of MMP9 was stimulated by IFN-α, tumor necrosis factor-α, and interleukin-1β. Remarkably, MMP-9 inhibition in IFN-γ-stimulated NS-SV-DC cells significantly decreased CXCL10 mRNA and secreted protein levels. Further analyses established that MMP-9 inhibition in IFN-γ-stimulated NS-SV-DC cells decreased STAT1 phosphorylation and hence suppressed IFN-γ signaling. Collectively, these results suggest that in addition to its reported role in the destruction of acinar structures, MMP-9 is involved in the IFN-γ-induced production of CXCL10 in pSS lesions. We believe that our findings open the door to the development of novel treatments for pSS, based on the modulation of MMP-9 activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Sjögren’s syndrome (SS), one of the most common autoimmune diseases [1, 2], is characterized by the destruction of the acinar structure by infiltrating mononuclear cells in the salivary and lacrimal glands [3,4,5]. Activated T and B cells constitute the majority of the infiltrating mononuclear cells in the lip salivary gland (LSG), while professional antigen-presenting cells, such as macrophages and dendritic cells (DCs), are correlated with the severity of the SS lesions [4, 5]. The mechanisms underlying the variability of glandular infiltration in SS salivary glands remain unclear. However, accumulating evidence indicates that several chemokines contribute to the development and progression of SS lesions [6,7,8,9].
C-X-C motif chemokine 10 (CXCL10) is a CXC chemokine induced by interferon (IFN)-γ and is produced by diverse cell types, including peripheral blood mononuclear cells, fibroblasts, and endothelial cells, during Th1-type immune responses [10]. Its cognate receptor C-X-C motif chemokine receptor 3 (CXCR3) is expressed on a wide range of cells of the innate immune system, including DCs, natural killer cells, natural killer T cells, neutrophils, and macrophages [11,12,13,14]. Therefore, these different cell types should be susceptible to CXCL10-mediated chemotaxis. Notably, gene expression profiling of LSG in control subjects and primary SS (pSS) patients has demonstrated that CXCL10 is upregulated in the latter [15]. Furthermore, CXCL10 is involved in the accumulation of infiltrating T cells in the salivary glands of pSS patients [7]. More recently, we reported that CXCL10 is secreted by ductal cells and induces CXCR3+ macrophage migration into the salivary glands of pSS patients [9].
Matrix metalloproteinases (MMPs) comprise a growing family of endopeptidases that can degrade extracellular matrix components, including collagens, elastin, laminins, and fibronectin, and thereby facilitate cell migration and tissue remodeling [16]. Importantly, the formation of normal acinar structures in the salivary glands depends on the integrity of the extracellular matrix, including the basement membrane [17, 18]. As is the case for CXCL10, MMP9 is over-expressed in the LSG of pSS patients [19]. Furthermore, previous studies suggest that increased MMP-9 activity and expression enhances the destruction of the basement membrane, which leads to the destruction of the acinar structures in SS salivary glands [20, 21]. Of note, recent studies also indicate that MMP-9 is important for the release of cytokines/chemokines [22, 23].
Collectively, these data support the notion that CXCL10 and MMP-9 both play roles in the pathogenesis of SS. However, the potential relationship between CXCL10 and MMP-9 has not been investigated. In this study, we used both LSG sections from pSS patients and human salivary gland cell lines to investigate the impact of MMP-9 activity on the production of CXCL10 in the salivary gland.
MATERIALS AND METHODS
Patients
Six female patients with pSS (mean age 64.2 ± 13.9 years) and three healthy female subjects (mean age 60.3 ± 4.2 years) were enrolled in this study. All individual participants included in the study were treated at Tokushima University Hospital between 2011 and 2016. The six pSS patients satisfied the revised Japanese Ministry of Health criteria for the diagnosis of SS [24] and the American College of Rheumatology classification criteria for SS [25]. The diagnosis of SS was based on the presence of two or more of the following clinicopathologic findings: lymphocytic infiltration of the salivary or lacrimal glands, dysfunction of salivary secretion, keratoconjunctivitis sicca, and presence of anti-Sjögren syndrome antigen A or B autoantibodies. The healthy subjects had experienced subjective symptoms of oral dryness but met none of the criteria for the diagnosis of SS.
Immunofluorescence Staining
Formalin-fixed paraffin-embedded LSG sections from pSS patients and healthy subjects were deparaffinized in xylene and rehydrated with graded ethanol (100%, 95%, 70%, and 50%). Antigen retrieval was performed by microwave treatment using a citrated-based antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s recommendations. Endogenous biotin was blocked using the Blocking One reagent (Nacalai Tesque, Kyoto, Japan) according to the manufacturer’s recommendations. Sections were then incubated overnight at 4 °C with rabbit polyclonal anti-human CXCL10 (Abcam, Cambridge, UK) and mouse monoclonal anti-human MMP-9 (Abcam) primary antibodies diluted 1:100 in PBS. After three washes with phosphate-buffered saline (PBS), the sections were incubated for 1 h at room temperature (RT) with goat anti-mouse IgG (H + L) conjugated to Alexa Fluor 488 secondary antibodies (Invitrogen, Carlsbad, CA, USA) diluted 1:200 in PBS. Next, the sections were washed in PBS and incubated for 1 h at RT with goat anti-rabbit IgG (H + L) conjugated to Alexa Fluor 568 secondary antibodies (Invitrogen) diluted 1:200. Finally, the nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI). The sections were observed using a Nikon A1 laser scanning confocal microscope (Nikon, Tokyo, Japan), and images were acquired using the NIS-Elements C Software (Nikon).
Cell Culture
The detailed characteristics of the immortalized normal human salivary gland ductal (NS-SV-DC) and acinar (NS-SV-AC) cell lines have been described elsewhere [26]. Both cell lines were cultured in keratinocyte serum-free medium (Gibco, Gaithersburg, MD, USA) in an incubator at 37 °C with a humidified atmosphere containing 5% CO2.
Reagents
Recombinant human IFN-α, IFN-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-1β were purchased from R&D Systems (Minneapolis, MN, USA). The MMP-9 inhibitor I was purchased from Merck (Darmstadt, Germany).
Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction
NS-SV-DC and NS-SV-AC cells were treated for 6, 12, or 24 h with 1000 IU/mL IFN-α, 10 ng/mL IFN-γ, 10 ng/mL TNF-α, and 1 ng/mL IL-1β. Where indicated, NS-SV-DC cells were treated for 6, 12, or 24 h with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. Total RNA was isolated using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s recommendations. Then, total RNA was converted into cDNA using the PrimeScript RT reagent Kit (TaKaRa, Kusatsu, Japan) according to the manufacturer’s recommendations. The mRNA levels of MMP9, CXCL10, IFN-γ receptor 1 (IFNGR1), IFNGR2, and GAPDH were analyzed using corresponding Assays-on-Demand Gene Expression Products (Applied Biosystems, Tokyo, Japan) and the TaqMan Universal PCR Master Mix (Applied Biosystems) with an ABI Prism 7000 Sequence Detection System (Applied Biosystems), according to the manufacturer’s recommendations. The thermal cycler conditions were 95 °C for 10 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Gene expression data were analyzed using the 2−ΔΔCT method of the Sequence Detection System software version 1 (Applied Biosystems). The relative quantification (RQ) of the fold change in gene expression was calculated using the formula RQ = 2−ΔΔCT and normalized using GAPDH as an internal reference. The relative mRNA levels are expressed as fold increase compared to the GAPDH mRNA level.
Reverse Transcription-Polymerase Chain Reaction
Specific custom sense and anti-sense primers for IFNGR1 (5′-GCTGTATGCCGAGATGGAAAA-3′ and 5′-AGGAAAATGGCTGGTATGACG-3′), IFNGR2 (5′-CGACAGTAAATGGTTCACGGC-3′ and 5′-TGGACATAATAACAAAAAAAGGC-3′), and GAPDH (5′-ACGCATTTGGCTGTATTGGG-3′ and 5′-TGATTTTGGAGGGATCTCGC-3′) were synthesized by Sigma (Deisenhofen, Germany). RT-PCR reactions were performed on a LifeECO Thermal Cycler (Nippon Genetics, Tokyo, Japan) using the Ex Taq DNA Polymerase (TaKaRa) according to the manufacturer’s recommendations. The thermal cycler conditions were 30 cycles of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 1 min. Amplification products were separated by 1.5% agarose gel electrophoresis and visualized after ethidium bromide staining using a FAS-III gel imaging system (TOYOBO, Osaka, Japan).
Enzyme-Linked Immunosorbent Assay
NS-SV-DC cells were incubated for 6, 12, or 24 h with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. The supernatants were collected, and the secreted CXCL10 protein levels were analyzed using a human CXCL10 ELISA kit (R&D Systems) according to the manufacturer’s recommendations. The absorbance at 450 nm was measured using a Multiskan JX microplate reader (Thermo Fisher Scientific, Waltham, MA, USA), and CXCL10 protein levels were determined using a standard curve.
Protein Isolation and Western Blot Analysis
NS-SV-DC cells were treated for 5, 30, 60, or 120 min with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. Whole-cell lysates were prepared using the M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) and Halt Protease Inhibitor Cocktail (Thermo Scientific) according to the manufacturer’s recommendations. Whole-cell lysate proteins (20 μg) were separated by electrophoresis using 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and then transferred onto nitrocellulose membranes. The membranes were blocked with 3% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) and incubated for 1 h at RT with anti-STAT1, anti-phospho-STAT1, anti-JAK2, anti-phospho-JAK2, and anti-β-actin primary antibodies (all from Cell Signaling Technology, Beverly, MA, USA) diluted 1:1000 in Can Get Signal Solution 1 (TOYOBO). After several washes with TBS-T, the membranes were incubated for 1 h at RT with appropriate secondary antibodies (Cell Signaling Technology) diluted 1:1000 in Can Get Signal Solution 2 (TOYOBO). The immune complexes were visualized using an ECL Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK). Densitometric analysis was performed using an Amersham Imager 600 (GE Healthcare) to determine the relative intensity of the immune complexes, using β-actin as an internal reference.
Statistical Analyses
All statistical analyses were performed using the SPSS Statistics version 15.0 software (IBM, Armonk, NY, USA). The data were analyzed using the non-parametric two-tailed Mann-Whitney U test, and statistical significance was defined as a p value < 0.05.
RESULTS
Expression of CXCL10 and MMP-9 in Lip Salivary Gland Sections from Sjögren’s Syndrome Patients
To assess the relationship between CXCL10 and MMP-9, we first used immunofluorescence staining to examine the expression level and localization of CXCL10 and MMP-9 in the LSG. Strikingly, CXCL10 and MMP-9 exhibited intense staining in the expanded ductal cells of pSS patients (Fig. 1a) compared to those of healthy subjects (Fig. 1b), which indicated increased expression of both proteins. Furthermore, inflammatory cells were detectable around the ductal structures of the salivary glands that showed CXCL10 and MMP-9 co-expression (Fig. 1a). The infiltrating inflammatory cells in the LSG of pSS patients did not show positive staining for CXCL10 or MMP-9 (Fig. 1a).

Histological analysis of LSG sections from pSS patients and healthy subjects. Representative images (from 3 to 5 samples) showing immunofluorescence staining for CXCL10 (green) and MMP-9 (red) in pSS patients (a) and healthy subjects (b). Nuclei (blue) were counterstained with DAPI. Arrowheads indicate inflammatory cells. Scale bars = 100 μm.
MMP-9 Expression Following IFN-α, IFN-γ, TNF-α, or IL-1β Stimulation of NS-SV-DC or NS-SV-AC Cells
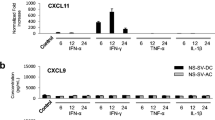
We have previously examined whether IFN-α, IFN-γ, TNF-α, and IL-1β could regulate the expression of CXCL10 mRNA in NS-SV-DC and NS-SV-AC cells [8]. According to our observation, IFN-γ-stimulated ductal cells were mainly responsible for CXCL10 overproduction in the salivary glands of pSS patients [8]. Therefore, to explore further the relationship between CXCL10 and MMP-9, we set out to identify factors that could regulate MMP-9 expression in human salivary gland cell lines. To this end, NS-SV-DC and NS-SV-AC cells were treated with IFN-α, IFN-γ, TNF-α, or IL-1β and MMP9 mRNA levels were examined by RT-qPCR. Remarkably, MMP9 expression in NS-SV-DC cells increased in response to IFN-α, TNF-α, and IL-1β stimulation but not following IFN-γ treatment (Fig. 2). In contrast, we could only detect a marginal increase in MMP9 mRNA levels in NS-SC-AC cells treated with IFN-γ, which suggested that MMP-9 was mainly produced by salivary gland ductal cells.

MMP9 expression in stimulated NS-SV-DC or NS-SV-AC cells. Histogram showing the relative changes in MMP9 mRNA levels in NS-SV-DC or NS-SV-AC cells treated for 6, 12, or 24 h with 1000 IU/mL IFN-α, 10 ng/mL IFN-γ, 10 ng/mL TNF-α, or 1 ng/mL IL-1β. Untreated cells were used as a control (C). Fold changes in mRNA levels were evaluated by RT-qPCR using GAPDH mRNA levels as an internal reference. Data are mean values ± standard deviation (SD) of three independent experiments.
Effects of MMP-9 Inhibition on CXCL10 Expression and Secretion in IFN-γ-Stimulated NS-SV-DC Cells
Next, we conducted RT-qPCR analyses to explore the potential involvement of MMP-9 in the regulation of CXCL10 expression. Remarkably, treatment with an MMP-9 inhibitor significantly decreased CXCL10 mRNA levels (p < 0.05) in IFN-γ-stimulated NS-SV-DC cells (Fig. 3), which suggested that MMP-9 activity was required for IFN-γ-induced CXCL10 expression.

Effect of MMP-9 inhibition on CXCL10 expression in IFN-γ-stimulated NS-SV-DC cells. Histogram showing the relative changes in CXCL10 mRNA levels in NS-SV-DC cells treated for 6, 12, or 24 h with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. Where indicated, cells were cultured in the absence of treatment (control) or with the MMP-9 inhibitor alone (MMP-9 inhibitor). Fold changes in mRNA levels were evaluated by RT-qPCR using GAPDH mRNA levels as an internal reference. Data are mean values ± SD of three independent experiments, and asterisks indicate statistical significance for the two-tailed Mann-Whitney U test (* p < 0.05).
To evaluate further the effects of MMP-9 inhibition, we performed an ELISA to measure secreted CXCL10 in the supernatants of NS-SV-DC cells treated with IFN-γ in the presence or absence of MMP-9 inhibitor. As anticipated, CXCL10 secretion by IFN-γ-stimulated NS-SV-DC was massively increased after 24 h (Fig. 4). In contrast, MMP-9 inhibition resulted in a significant decrease in the production of CXCL10 after 24 h (p < 0.05), which indicated that MMP-9 activity supported IFN-γ-induced CXCL10 secretion by salivary gland ductal cells.

Effect of MMP-9 inhibition on CXCL10 secretion by the IFN-γ-stimulated NS-SV-DC cells. Histogram showing the concentration of CXCL10 measured by ELISA in the supernatants of NS-SV-DC cells treated for 6, 12, or 24 h with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. Where indicated, cells were cultured in the absence of treatment (control) or with the MMP-9 inhibitor alone (MMP-9 inhibitor). Data are mean values ± SD of three independent experiments, and asterisks indicate statistical significance for the two-tailed Mann-Whitney U test (* p < 0.05).
Effects of MMP-9 Inhibition on IFN-γ Receptor Expression in NS-SV-DC Cells
Since MMP-9 inhibition suppressed CXCL10 both at the mRNA and protein levels, we hypothesized that the MMP-9 inhibitor could affect the IFN-γ receptors (IFNGRs). IFNGRs consist of two subunits: IFNGR1 (also known as the IFN-γ receptor α chain) and IFNGR2 (also known as the IFN-γ receptor β chain) [27]. We first used RT-PCR to examine the expression of IFNGR1 and IFNGR2 in NS-SV-DC cells and found no perceptible difference in the mRNA levels following IFN-γ stimulation and MMP-9 inhibition (Fig. 5a). Further analyses using RT-qPCR confirmed this observation (Fig. 5b), which indicated that MMP-9 inhibition did not significantly affect the mRNA levels of IFNGR1 and IFNGR2.

Effects of MMP-9 inhibition on IFN-γ receptors expression in NS-SV-DC cells. a RT-PCR products separated by agarose gel electrophoresis showing the mRNAs levels of IFNGR1 and IFNGR2. GAPDH was used as an internal control. Where indicated, NS-SV-DC cells were treated with 10 ng/mL IFN-γ and 5 μM MMP-9 inhibitor. b Histogram showing the relative changes in IFNGR1 and IFNGR2 mRNA levels in NS-SV-DC cells treated for 6, 12, or 24 h with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. Where indicated, cells were cultured in the absence of treatment (control) or with the MMP-9 inhibitor alone (MMP-9 inhibitor). Fold changes in mRNA levels were evaluated by RT-qPCR using GAPDH mRNA levels as an internal reference. Data are mean values ± SD of three independent experiments.
Effects of MMP-9 Inhibition on the IFN-γ Signaling Pathway in NS-SV-DC Cells
We previously reported that IFN-γ stimulated the production of CXCL10 via the JAK2/STAT1 signaling pathway [8]. To dissect the molecular mechanisms underlying the effects of MMP-9 inhibition, we thus performed western blot analyses to assess the expression levels of STAT1 and JAK2 and their phosphorylation, which denote STAT1 and JAK2 activation. We did not examine JAK1 since our previous report suggested that it was not expressed in NS-SV-DC cells [8]. Whether cells were treated or not with the MMP-9 inhibitor, we did not observe any changes in JAK2 levels and phosphorylation upon IFN-γ stimulation (Fig. 6a). In striking contrast, we found that STAT1 phosphorylation was both delayed and reduced following MMP-9 inhibition in IFN-γ-stimulated NS-SV-DC cells (Fig. 6), which indicated that MMP-9 activity was involved in STAT1-mediated IFN-γ signaling.

Effects of MMP-9 inhibition on IFN-γ-induced STAT1 and JAK2 phosphorylation in NS-SV-DC cells. a Representative western blot showing STAT1, JAK2, phosphorylated-STAT1 (p-STAT1), and phosphorylated-JAK2 (p-JAK2) protein levels in NS-SV-DC cells treated for 5, 30, 60, or 120 min with 10 ng/mL IFN-γ in the presence or absence of 5 μM MMP-9 inhibitor. β-actin was used as a loading control. b Histogram showing the relative STAT1 phosphorylation levels as determined by densitometric analysis of the western blot presented in panel a. The intensity of the p-STAT1 band was normalized to the intensity of the corresponding STAT1 and β-actin bands.
DISCUSSION
Although previous studies reported that CXCL10 and MMP9 were both upregulated in the LSG of pSS patients [15, 19] and MMP-9 was important for the release of cytokines/chemokines [22, 23], there is, to the best of our knowledge, no study investigating the potential relationship between CXCL10 and MMP-9 in SS. Therefore, in this study, we investigated the functional relationship between CXCL10 and MMP-9 using LSG sections from pSS patients and human salivary gland cell lines.
SS is a chronic autoimmune disease characterized by the destruction of the salivary and lacrimal glands [1,2,3]. Histologically, pSS patients exhibit selective and progressive destruction of the acinar structures in the LSG, which is concomitant with the infiltration of various mononuclear cells, including T and B cells, DCs, and macrophages [4, 5]. Although the underlying molecular mechanisms leading to mononuclear cell infiltration in the salivary glands of pSS patients remain elusive, accumulating evidence indicates that the cytokines-chemokine-driven network is one of the key processes in the autoimmune response associated with pSS [6,7,8,9, 28].
In particular, CXCL10 overexpression has been observed in the ductal epithelium of the LSG of pSS patients [7, 9]. Importantly, this increased production of CXCL10 by the ductal epithelium regulated immune responses through the recruitment of CXCR3+ mononuclear cells, including T cells, DCs, and macrophages [7, 9]. The involvement of MMP-9 in the pathogenesis of pSS derives from the observation that increased MMP-9 expression and activity results in the disruption of the basement membrane, which in turn leads to the destruction of the acinar structures in SS salivary glands [22, 23].
Our immunofluorescence analyses in LSG sections from pSS patients showed that CXCL10 and MMP-9 were strongly co-expressed in expanded ductal cells, which was associated with the presence of infiltrating immune cells around these expanded ductal cells. In contrast, CXCL10 and MMP-9 expression was not detected in acinar cells (Fig. 1). A previous study reported that CXCL10 increases MMP-9 secretion from monocytes and neutrophils but not from lymphocytes in patients with bullous pemphigoid, the most common autoimmune subepidermal blistering disease [29]. In contrast, our findings suggested that most of the infiltrating immune cells were negative for MMP-9 staining. However, recent studies highlighted the role of MMP-9 in the release of cytokines/chemokines [22, 23]. Since we observed numerous infiltrating immune cells around the ductal structures that co-expressed CXCL10 and MMP-9, we speculated that MMP-9 could be involved in the secretion of CXCL10 by ductal cells and therefore sought to identify the factors that could regulate MMP-9 expression in human salivary gland ductal and acinar cells.
The promoter region of the human MMP9 gene includes two AP-1 binding sites, as well as NF-κB, SP-1, GT box, and PEA3 elements [30], which mediate transcriptional regulation by various factors, such as TNF-α, IL-1β, PMA, v-src, ras, and LPS. Furthermore, IFN-γ has been shown to suppress MMP9 expression through the inhibition of transcription factors, including STAT-1α, CREB, and AP-1 [30, 31]. While our RT-qPCR data showing that MMP9 expression did not increase in IFN-γ stimulated NS-SV-DC ductal cells are consistent with these reports, we also found that MMP9 expression was markedly higher in the NS-SV-DC ductal cell line than in the NS-SV-AC acinar cell line. Moreover, we demonstrated that MMP9 expression in NS-SV-DC cells increased following IFN-α, TNF-α, or IL-1β stimulation, providing new insight into the regulation of MMP-9 in the salivary glands.
MMPs are known to cleave several pro-inflammatory chemokines, thereby modulating their function and having an impact on the inflammatory process [22, 28]. Importantly, MMP-2 and MMP-9 can cleave CXCL10, which reduces its chemotactic potential through the removal of N-terminal residues [32]. Here, we demonstrated that inhibition of MMP-9 activity suppressed IFN-γ-induced CXCL10 at both mRNA and secreted protein levels. Although we expected MMP-9 to be involved in CXCL10 secretion by NS-SV-DC ductal cells, the observation that MMP-9 inhibition affected CXCL10 mRNA transcription was surprising. Indeed, our data on IFN-γ signaling established that MMP-9 was not only involved in the cleavage of CXCL10 but also in the activation of STAT1, which is known to mediate IFN-γ-induced CXCL10 expression.
Based on the evidence available, the progression of pSS is proposed to take place in three phases. The first phase corresponds to a preexisting condition characterized by genetic susceptibility factors. The second is a lymphocyte-independent phase that occurs following the release of antigens by necrotic and apoptotic cells or viral trigger, which elicit an innate immune response with cytokine production. The third phase is a tissue-specific immunological attack by activated T and B cells, and their products, against corresponding self-antigens, which leads to autoimmune exocrinopathy [19, 33]. In the second and third phases, salivary gland cells, particularly ductal cells, likely produce CXCL10 and MMP-9. Collectively, our findings support the notion that MMP-9 could promote the secretion of CXCL10 by ductal cells in SS salivary glands via IFN-γ signaling and the activation of STAT1.
In conclusion, this study provides new insight into the underlying molecular mechanisms of SS by demonstrating that MMP-9 inhibition could suppress CXCL10 expression in human salivary gland ductal cells via a decrease in STAT1 phosphorylation and, therefore, IFN-γ signaling. We would like to propose that in addition to its role in the destruction of acinar structures through the degradation of the basement membrane, MMP-9 also contributes to the pathogenesis of pSS by supporting the IFN-γ-induced production of CXCL10 in pSS lesions. Therefore, we believe that our data provide a strong theoretical basis for the development of novel approaches for the treatment of pSS, based on the modulation of MMP-9 activity.
References
Alspaugh, M.A., and K. Whaley. 1981. Sjögren’s syndrome. In Textbook of Rheumatology, ed. W.N. Kelley, E.D. Harris, S. Ruddy, and C.B. Sledge, 971–999. Philadelphia: Saunders (imprint).
Vivino, F.B., V.Y. Bunya, G. Massaro-Giordano, C.R. Johr, S.L. Giattino, A. Schorpion, B. Shafer, A. Peck, K. Sivils, A. Rasmussen, J.A. Chiorini, J. He, and J.L. Ambrus Jr. 2019. Sjogren’s syndrome: An update on disease pathogenesis, clinical manifestations and treatment. Clinical Immunology 203: 81–121.
Daniel, T.E. 1984. Labial salivary gland biopsy in Sjögren’s syndrome. Assessment as a diagnostic criterion in 362 suspected cases. Arthritis and Rheumatism 27: 147–156.
Christodoulou, M.I., E.K. Kapsogeorgou, and H.M. Moutsopoulos. 2010. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. Journal of Autoimmunity 34: 400–407.
Kamiński, B. 2019. Laryngological manifestations of Sjögren’s syndrome. Reumatologia 57: 37–44.
Lee, Y.J., R.H. Scofield, J.Y. Hyon, P.Y. Yun, H.J. Lee, E.Y. Lee, E.B. Lee, and Y.W. Song. 2010. Salivary chemokine levels in patients with primary Sjögren’s syndrome. Rheumatology (Oxford) 49: 1747–1752.
Ogawa, N., L. Ping, L. Zhenjun, Y. Takada, and S. Sugai. 2002. Involvement of the interferon-γ-induced T cell-attracting chemokines, interferon-γ-inducible 10-kd protein (CXCL10) and monokine induced by interferon-γ (CXCL9), in the salivary gland lesions of patients with Sjögren’s syndrome. Arthritis and Rheumatism 46: 2730–2741.
Aota, K., K. Kani, T. Yamanoi, K.I. Nakashiro, N. Ishimaru, and M. Azuma. 2018. Distinct regulation of CXCL10 production by cytokines in human salivary gland ductal and acinar cells. Inflammation 41: 1172–1181.
Aota, K., T. Yamanoi, K. Kani, K.I. Nakashiro, N. Ishimaru, and M. Azuma. 2018. Inverse correlation between the number of CXCR3+ macrophages and the severity of inflammatory lesions in Sjögren's syndrome salivary glands: A pilot study. Journal of Oral Pathology and Medicine 47: 710–718.
Luster, A.D., J.C. Unkeless, and J.V. Ravetch. 1985. γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 315: 672–676.
García-López, M.A., F. Sánchez-Madrid, J.M. Rodríguez-Frade, M. Mellado, A. Acevedo, M.I. García, J.P. Albar, C. Martínez-A, and M. Marazuela. 2001. CXCR3 chemokine receptor distribution in normal and inflamed tissues: Expression on activated lymphocytes, endothelial cells, and dendritic cells. Laboratory Investigation 81: 409–418.
Groom, J.R., and A.D. Luster. 2011. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunology and Cell Biology 89: 207–215.
Hartl, D., S. Krauss-Etschmann, B. Koller, P.L. Hordijk, T.W. Kuijpers, F. Hoffmann, A. Hector, E. Eber, V. Marcos, I. Bittmann, O. Eickelberg, M. Griese, and D. Roos. 2008. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. The Journal of Immunology 181: 8053–8067.
Tomita, K., B.L. Freeman, S.F. Bronk, N.K. LeBrasseur, T.A. White, P. Hirsova, and S.H. Ibrahim. 2016. CXCL10-mediates macrophage, but not other innate immune cells-associated inflammation in murine nonalcoholic steatohepatitis. Scientific Reports 6: 28786.
Hjelmervik, T.O., K. Petersen, I. Jonassen, R. Jonsson, and A.I. Bolstad. 2005. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren’s syndrome patients from healthy control subjects. Arthritis and Rheumatism 52: 1534–1544.
Szabo, K.A., R.J. Ablin, and G. Singh. 2004. Matrix metalloproteinases and the immune response. Clinical and Applied Immunology Reviews 4: 295–319.
Bernfield, M.R., S.D. Banerjee, and R.H. Cohn. 1972. Dependence of salivary epithelial morphology and branching morphogenesis upon acid mucopolysaccharide-protein (proteoglycan) at the epithelial surface. Journal of Cell Biology 52: 674–689.
Patel, V.N., and M.P. Hoffman. 2014. Salivary gland development: A template for regeneration. Seminars in Cell and Developmental Biology 25-26: 52–60.
Pérez, P., J.M. Anaya, S. Aguilera, U. Urzúa, D. Munroe, C. Molina, M.A. Hermoso, J.M. Cherry, C. Alliende, N. Olea, E. Ruiz-Narváez, and M.J. González. 2009. Gene expression and chromosomal location for susceptibility to Sjögren’s syndrome. Journal of Autoimmunity 33: 99–108.
Konttinen, Y.T., S. Halinen, R. Hanemaaijer, T. Sorsa, J. Hietanen, A. Ceponis, J.W. Xu, R. Manthorpe, J. Whittington, A. Larsson, T. Salo, L. Kjeldsen, U.H. Stenman, and A.Z. Eisen. 1998. Matrix metalloproteinase (MMP)-9 type IV collagenase/gelatinase implicated in the pathogenesis of Sjögren’s syndrome. Matrix Biology 17: 335–347.
Azuma, M., K. Aota, T. Tamatani, K. Motegi, T. Yamashita, K. Harada, Y. Hayashi, and M. Sato. 2000. Suppression of tumor necrosis factor α-induced matrix metalloproteinase 9 production by the introduction of a super-repressor form of inhibitor of nuclear factor κBα complementary DNA into immortalized human salivary gland acinar cells: Prevention of the destruction of the acinar structure in Sjögren's syndrome salivary glands. Arthritis and Rheumatism 43: 1756–1767.
Song, J., C. Wu, X. Zhang, and L.M. Sorokin. 2013. In vivo processing of CXCL5 (LIX) by matrix metalloproteinase (MMP)-2 and MMP-9 promotes early neutrophil recruitment in IL-1β-induced peritonitis. The Journal of Immunology 190: 401–410.
Munakata, S., Y. Tashiro, C. Nishida, A. Sato, H. Komiyama, H. Shimazu, D. Dhahri, Y. Salama, S. Eiamboonsert, K. Takeda, H. Yagita, Y. Tsuda, Y. Okada, H. Nakauchi, K. Sakamoto, B. Heissig, and K. Hattori. 2015. Inhibition of plasmin protects against colitis in mice by suppressing matrix metalloproteinase 9-mediated cytokine release from myeloid cells. Gastroenterology 148: 565–578.
Fujibayashi, T., S. Sugai, N. Miyasaka, Y. Hayashi, and K. Tsubota. 2004. Revised Japanese criteria for Sjögren's syndrome (1999): Availability and validity. Modern Rheumatology 14: 425–434.
Shiboski, S.C., C.H. Shiboski, L.A. Criswell, A.N. Baer, S. Challacombe, H. Lanfranchi, M. Schiødt, H. Umehara, F. Vivino, Y. Zhao, Y. Dong, D. Greenspan, A.M. Heidenreich, P. Helin, B. Kirkham, K. Kitagawa, G. Larkin, M. Li, T. Lietman, J. Lindegaard, N. McNamara, K. Sack, P. Shirlaw, S. Sugai, C. Vollenweider, J. Whitcher, A. Wu, S. Zhang, W. Zhang, J.S. Greenspan, and T.E. Daniels for the Sjögren’s International Collaborative Clinical Alliance (SICCA) Research Groups. 2012. American College of Rheumatology classification criteria for Sjögren's syndrome: A data-driven, expert consensus approach in the Sjögren’s International Collaborative Clinical Alliance cohort. Arthritis Care and Research 64: 475–487.
Azuma, M., T. Tamatani, Y. Kasai, and M. Sato. 1993. Immortalization of normal human salivary gland cells with duct-, myoepithelial-, acinar-, or squamous phenotype by transfection with SV40 ori- mutant deoxyribonucleic acid. Laboratory Investigation 69: 24–42.
Bach, E.A., M. Aguet, and R.D. Schreiber. 1997. The IFNγ receptor: A paradigm for cytokine receptor signaling. Annual Review of Immunology 15: 563–591.
Van Lint, P., and C. Libert. 2007. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. Journal of Leukocyte Biology 82: 1375–1381.
Riani, M., S. Le Jan, J. Plée, A. Durlach, R. Le Naour, G. Haegeman, P. Bernard, and F. Antonicelli. 2017. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. The Journal of Allergy and Clinical Immunology 139: 863–872.
Ma, Z., H. Qin, and E.N. Benveniste. 2001. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-γ and IFN-β: Critical role of STAT-1α. The Journal of Immunology 167: 5150–5159.
Hu, X., and L.B. Ivashkiv. 2009. Cross-regulation of signaling pathways by interferon-γ: Implications for immune responses and autoimmune diseases. Immunity 31: 539–550.
Proost, P., E. Schutyser, P. Menten, S. Struyf, A. Wuyts, G. Opdenakker, M. Detheux, M. Parmentier, C. Durinx, A.M. Lambeir, J. Neyts, S. Liekens, P.C. Maudgal, A. Billiau, and J. Van Damme. 2001. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 98: 3554–3561.
Mitsias, D.I., E.K. Kapsogeorgou, and H.M. Moutsopoulos. 2006. The role of epithelial cells in the initiation and perpetuation of autoimmune lesions: Lessons from Sjogren’s syndrome (autoimmune epithelitis). Lupus 15: 255–261.
Funding
This work was supported by the Grants-in-Aid for Scientific Research program from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (No. 19 K10311).
Author information
Authors and Affiliations
Contributions
KA and MA contributed to the study conception and design. Material preparation, data collection, and analysis were performed by KA, SO, TY, KK, and YM. The first draft of the manuscript was written by KA, and all authors commented on previous versions of the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Board of Tokushima University Hospital (no. 2802) and with the 1964 Helsinki declaration and its later amendments.
Informed Consent
Written informed consent was obtained from all individual participants included in the study, and this process was documented by the Institutional Review Board of Tokushima University Hospital. The informed consent procedure was approved by the Ethics Committee of Tokushima University Hospital.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aota, K., Ono, S., Yamanoi, T. et al. MMP-9 Inhibition Suppresses Interferon-γ-Induced CXCL10 Production in Human Salivary Gland Ductal Cells. Inflammation 42, 2148–2158 (2019). https://doi.org/10.1007/s10753-019-01079-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-019-01079-x