
Abstract
Asbestos and silica (exogenous danger) and adenosine triphosphate (ATP, endogenous danger-signaling molecule) synergistically increase IL-1β release from endotoxin-primed macrophage, which is mediated by NOD-like receptor protein 3 (NLRP3) inflammasome. However, the conversion of pro-IL-1β to its active form seems to depend on the macrophage cell types. In the present study, bone marrow-derived macrophages (BMM) and three murine macrophage cell lines, J774.1, J774A.1, and RAW264.7 were exposed to ATP or fibrous titanium dioxide (FTiO2) in the presence or absence of lipopolysaccharide (LPS), and the concentrations of IL-1β and IL-6 in both cell lysates and in the culture media were measured by immunoblotting to differentiate active form of IL-1β from pro-IL-1β. IL-1β release was synergistically increased when the cells were exposed to both LPS and ATP or FTiO2, while IL-6 was readily released by LPS alone. IL-1β released into the culture medium was pro-IL-1β in J774.1 and RAW264.7, and most of the pro-IL-1β remained inside the cells. In contrast, the active form of IL-1β was released together with pro-IL-1β from J774A.1 and BMM after the co-stimulation. J774A.1 and BMM express apoptosis-associated speck-like protein contains a carboxyl-terminal CARD (ASC) while J774.1 and RAW264.7 do not or only faintly express ASC, and accordingly, caspase-1, which converts pro-IL-1β to its active form, is activated only in J774A.1 and BMM. Collectively, the canonical inflammasome pathway is not activated in J774.1 and RAW264.7, and the apparent synergistical increase of IL-1β in the culture medium mostly reflects the leakage of pro-IL-1β from these cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Alveolar macrophages are host defense cells against both inhaled environmental particulate substances and microorganisms. Endotoxin is an outer membrane component of gram-negative bacteria and is ubiquitous even in the atmosphere [1]. Several lines of evidence indicate that particulate substances synergistically enhance inflammatory responses of macrophages to endotoxin [2]. However, the synergistic mechanism was not well understood until recently.
Over the last decade, inflammasome studies have shed light on the synergistic effects of particulate substances and endotoxin on macrophage-mediated inflammatory responses. Compared to non-exposed cells, lipopolysaccharide (LPS)-primed mouse bone marrow-derived macrophages (BMM), human monocyte-derived macrophages, and THP-1 cells (a human leukemic monocyte cell line) release much more IL-1β when exposed to particulate substances such as asbestos, silica, and monosodium urate but not when exposed to diesel exhaust particulates or cigarette smoke extract [3]. In the canonical inflammasome pathway, LPS increases intracellular pro-IL-1β via the NF-κB pathway, and fibrous or crystalline particles injure the lysosomal membrane, thereby inducing the formation of an apoptosis-associated speck-like protein containing a carboxyl-terminal CARD (ASC) speckle and activating pro-caspase-1. Finally, mature IL-1β (17 kDa) is processed from pro-IL-1β by cleaved and active caspase-1 [4, 5].
However, there are contradictory data regarding the canonical inflammasome pathway from both cell biological and toxicological points of view. Mature IL-1β is released after cleavage by caspase-1 although IL-1β lacks the amino-acid sequence that would direct it to the Golgi apparatus, an organelle that mediates protein secretion [6]. Nano-TiO2 and nano-SiO2, but not nano-ZnO, activated the NLR pyrin domain containing three (NLRP3) inflammasomes that lead to IL-1β release [7]. Fumed (non-crystalline) silica particles induced almost the same IL-1β response as crystalline silica in RAW264.7 and rat lung primary macrophages, suggesting that crystallinity may not be a major factor for induction of the inflammasome [2]. Exposure to 20- and 1000-nm latex beads resulted in IL-1β release, but 200-nm beads did not induce IL-1β production. IL-1β production induced by 20-nm latex beads was mediated via cathepsin B leakage from ruptured phagolysosomes, and IL-1β production by 1000-nm latex beads depended on ROS production in LPS-primed (co-exposed) mouse BMM [8].
It has been reported that various particulate substances such as silver nanoparticles [9], asbestos, and multi-walled carbon nanotubes [10] activate caspase-1 and increase IL-1β production in LPS-primed THP-1 cells. It is of interest to note that long fibrous TiO2 (FTiO2) but not spherical or short TiO2 induced canonical inflammasome responses such as IL-1β and IL-18 production in mouse alveolar macrophages in a manner very similar to asbestos or silica when co-stimulated with LPS [11]. The authors reported that the same effects were observed in THP-1 human macrophages, but not in RAW or MH-S murine macrophages. Pro-IL-1β was the predominant form of extracellular IL-1β in cultured macrophages, whereas monocytes preferentially released the 17-kDa mature IL-1β when human alveolar macrophages and autologous peripheral blood monocytes were stimulated with LPS [12]. Moreover, differential patterns of IL-1 production and secretion were observed between bone marrow-derived dendritic cells (BMDC) and BMM; BMM were more sensitive to LPS than BMDC and produced IL-1β intracellularly, whereas BMDC readily secreted IL-1β [13]. The active form of caspase-1 and mature IL-1β (17 kDa) was not formed following stimulation of LPS-primed RAW264.7 cells with ATP because this cell line, which has functional P2X7 purinergic receptors, lacks ASC. In contrast, J774A.1 cells and mouse peritoneal macrophages responded to ATP via the inflammasome pathway. There may be an as-yet-undiscovered mechanism leading to the release of pro-IL-1β, because pro-IL-1β was released into the culture medium in ATP-stimulated RAW264.7 cells [14].
Those previous reports suggest a large difference in IL-1β production among innate immune cells and led us to investigate whether co-exposure of BMM or murine macrophage cell lines such as J774.1, J774A.1, and RAW264.7 to LPS and fibrous TiO2 or ATP causes canonical inflammasome activation and enhances IL-1β release. We report that the IL-1β-release patterns in J774.1 and RAW264.7 murine cell lines are different from those in BMM and J774A.1 cells. We also report that an increase in pro-IL-1β messenger RNA and a leakage of pro-IL-1β are responsible for an apparent synergistic increase in IL-1β release in J774.1 cells following co-stimulation with LPS and FTiO2 or ATP.
MATERIALS AND METHODS
Chemicals
The following chemicals were used: WST-8 Cell Counting kit (Dojindo, Osaka, Japan); caspase-1 fluorometric assay kits, z-VAD-FMK and z-YVAD-FMK (BioVision, Milpitas, CA); NuPAGE® and LDS (lithium dodecyl sulfate) sample buffer, iBlot® polyvinylidene difluoride (PVDF), Opti-MEM culture medium (Life Technology-Invitrogen, Carlsbad, CA); radio immunoprecipitation assay (RIPA) buffer with protease inhibitors, anti-caspase-1 p10 antibody (M-20), and POD-conjugated anti-mouse or rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA); phosphatase inhibitor cocktail (Thermo Fischer Scientific, Waltham, MA); PureLink RNA mini kit with Trizol and DNase I (Life Technology-Ambion, Austin, TX); POD-conjugated anti-GAPDH antibody (MBL, Nagoya, Japan); anti-IL-1β (rabbit mAb, D3H1Z), anti-IL-6 (rabbit mAb, D5W4V), anti-ASC (rabbit mAb, D2W8U), and anti-NLPR3 antibodies (rabbit mAb, D4D8T) (Cell Signaling Technology, Danvers, MA); ECL prime western detection reagent (HE Healthcare, Buckinghamshire, UK); PVDF blocking reagent and Can-Get-Signal™ (TOYOBO, Osaka, Japan); PrimeScript™ RT reagent kit and SYBR® Premix Ex Taq™ II (TaKaRa, Tokyo, Japan); M-CSF (Wako, Osaka, Japan); adenosine and adenosine 5′-triphosphate (ATP) disodium salt (Sigma, St. Louis, MO). Other common buffers and reagents were of analytical or biochemical grade.
Particulate Substances
Fibrous titanium dioxide (FTiO2, #TO1) was obtained from the Japan Fibrous Material Research Association (JFMRA, Tokyo, Japan). The nominal average length and width of FTiO2 were 2.1 and 0.14 μm, respectively. Tricalcium phosphate (TCP, <100 nm) was purchased from Sigma-Aldrich (St. Louis, MO). These particles were heat-treated (250 °C, 2 h) in an electric furnace to remove potentially contaminating endotoxin and were suspended in PBS or culture medium by sonication before use.
Cells
J774.1 (RBC0434), J774A.1 (JCRB9108), and RAW264.7 murine macrophages (TIB-71) were obtained from the RIKEN Institute (Tsukuba, Japan), NIBIOHN (Tokyo, Japan), and the ATCC (Manassas, VA), respectively. Unless otherwise specified, J774.1 and J774A.1 cells were cultured in RPIM1640 and RAW264.7 cells in Dulbecco’s modified Eagle medium (DMEM) containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. BMM were obtained from male-specific pathogen-free C57BL/6-J mice. The mice were purchased from SLC Japan (Shizuoka, Japan). At the age of 6 weeks, the animals were injected (i.p.) with an anesthetic combination of 0.75 mg/kg medetomidine hydrochloride, 4.0 mg/kg midazolam, and 5.0 mg/kg butorphanol tartrate and were exsanguinated from the abdominal aorta under anesthesia. One right femoral bone was obtained from each of five animals. The bone marrow was flushed out by phosphate buffered saline (PBS), and the cells were cultured in complete RPMI1640 culture medium containing 10 ng/mL M-CSF. The medium was changed on days 3 and 5. After 7 days of culture, the cells were used for experiments as BMM. These procedures were approved by the Animal Care and Use Committee of the National Institute for Environmental Studies (NIES).
Cell Viability
J774.1, J774A.1, or RAW264.7 cells were suspended in complete RPMI1640 medium at a density of 0.15 × 106 cells/mL and a 100-μL aliquot was placed in a 96-well culture dish. The cells were pre-cultured for 24 h prior to exposure to test particles or chemicals. The cells were washed twice with Hank’s balanced salt solution (HBSS), and the number of viable cells was evaluated by a modified MTT assay method using the WST-8 Cell Counting kit. The reaction was stopped by adding one tenth volume of 0.1 mol/L HCl solution after the development of chromophore. The O.D. at 450 nm was measured using a microplate reader (POLARstar OPTIMA, BMG Labtech, Offenburg, Germany).
Measurement of Caspase-1 Activity
J774.1 cells cultured in a 12-well culture dish were exposed to 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 24 h. The cells were then lysed with 80 μL of lysis buffer, and the lysate was centrifuged at 9000×g for 10 min at 4 °C. A 40-μL aliquot of the supernatant was mixed and incubated with the substrate at 37 °C with intermittent vortexing for 1 h. The fluorescence intensity (Ex: 410 nm, Em: 520 nm) was measured using a microplate reader.
Cytokine Array
The culture medium obtained from J774.1 cells was centrifuged at 600×g for 10 min at 4 °C, and the supernatant was collected. Concentrations of cytokines in the culture medium were measured using an antibody array (Proteome Profiler, Mouse cytokine array, R&D, Minneapolis, MN) according to the manufacturer’s instructions. The array membrane was illuminated with ECL, and cytokine levels were quantified using a chemiluminescence densitometer (Lumino Imaging Analyzer, FAS-1100, TOYOBO, Osaka, Japan).
Quantitative IL-1β Measurement
Concentrations of IL-1β in the culture medium were measured using a mouse IL-1β ELISA kit (Thermo Fischer Scientific) according to the manufacturer’s instructions. The recovery of IL-1β from the culture medium was also measured using mouse recombinant IL-1β that was included in the kit as a standard.
Immunoblot Analysis
Cell monolayers were rinsed twice with PBS and lysed on ice for 10 min with cold RIPA lysis buffer (1 × TBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing protease and phosphatase inhibitor cocktails. Each lysate was transferred to a conical tube and centrifuged at 9000×g for 5 min at 4 °C. Aliquots of the supernatant were mixed with LDS sample buffer (1 × TBS, 10% glycerol, 0.015% EDTA, 50 mM DTT, and 2% LDS) and heated at 95 °C for 5 min. Proteins were separated on 4–12% gels by SDS-PAGE and then electroblotted onto a PVDF membrane. The membrane was blocked with a non-protein blocking agent and reacted with primary antibody for 1 h. Primary (1:200–500 dilution) and secondary (1:2000 dilution) antibodies were dissolved in Can-Get-Signal® solution. The antibodies were removed by incubating each membrane in stripping buffer containing 2% SDS and 100 mM 2-mercaptoethanol at 56 °C for 15 min, and the membrane was re-probed with anti-GAPDH antibody to normalize the protein loading level. The culture medium was first centrifuged at 120 g for 5 min to remove cell debris, and the clear supernatant was concentrated 50 times before SDS-PAGE electrophoresis by ultrafiltration using an Amicon Ultra (MW 3000, Millipore, Cork, Ireland).
Quantitative Real Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
IL-1β and IL-6 mRNA levels in J774.1 cells were measured using a thermal cycler (TP800, Takara Bio, Otsu, Japan) adopting the ∆∆Ct method. Briefly, total RNA was extracted from the cells using the PureLink RNA Mini kit with Trizol and on-column DNase-treatment. Complementary DNA was prepared using a PrimeScript RT reagent kit. PCR reactions were performed using SYBR Premix Ex Taq II and the following gene specific primers:
-
IL-1β forward, 5′-CCAGCTTCAAATCTCACAGCAG-3′
-
IL-1β reverse, 5′-CTTCTTTGGGTATTGCTTGGGATC-3′
-
IL-6 forward, 5′-TCCAGTTGCCTTCTTGGGAC-3′
-
IL-6 reverse, 5′-GTACTCCAGAAGACCAGAGG-3′
-
Gapdh forward, 5′- AACGACCCCTTCATTGAC-3′
-
Gapdh reverse, 5′- TCCACGACATACTCAGCAC-3′
Statistical Analyses
Data are presented as means ± SEM. One-way or two-way ANOVA was applied first and, when significant differences were observed, Tukey’s post-hoc comparison was made. Probability values less than 0.05 were accepted as indicative of statistical significance.
RESULTS
First, we investigated inflammatory responses of three murine macrophage cell lines to ATP after pre-stimulation with LPS. J774A.1, J774.1, and RAW264.7 cells were exposed to ATP, LPS, and LPS plus ATP, and the amounts of IL-1β, IL-6, and caspase-1 in both the cell lysate and culture medium were measured by immunoblot analyses (Fig. 1). ATP caused acute cytotoxic effects on these macrophages at the concentration of 2 mM or higher, while 2–5 mM adenosine was not cytotoxic (Fig. 1c). J774A.1 cells seemed to respond to LPS with ATP in the canonical inflammasome pathway, since pro-caspase-1 (45 kDa) was converted to the active form (10 kDa), and the cleaved and active form of IL-1β (17 kDa) was released in to the culture medium (Fig. 1a). In contrast, neither pro-caspase-1 nor pro-IL-1β (31 kDa) was cleaved in J774.1 and RAW264.7 cells, and most pro-caspase-1 and pro-IL-1β remained inside the cells (Fig. 1b). The high intensification detected a putative processed IL-1β at 21 kDa in the cell lysate of RAW264.7 cells (Supplementary Fig. 1). However, its molecular weight was different from the active form of IL-1β (17 kDa). IL-6 was released in response to LPS alone, and ATP appeared to decrease the production of IL-6 in all the three cell types.

Immunoblot analysis for the detection of IL-1β, IL-6, and caspase-1 in the cell lysate and the culture medium of J774A.1 (a) and J774.1 and RAW264.7 cells (b), and cytotoxic effects of ATP (c). (a, b) The cells were exposed to 5 mM ATP for the indicated time alone or at the end of 6-h exposure to 100 ng/mL of LPS. Prior to analysis, the culture medium was centrifuged to remove cell debris, and the supernatant was concentrated 50 times using an ultrafiltration membrane (MW, 3000). The cells in a 60-mm diameter dish were lysed with 100-μL RIPA buffer. Cleaved IL-1β and cleaved caspase-1 were visible in neither the cell lysate nor culture medium of J774.1 and RAW264.7 cell. (c) J774.1 (open column), J774A.1 (crosshatched column), and RAW264.7 cells (gray column) were exposed to the indicated concentrations of ATP or adenosine for 4 h, following which cell viability was assayed using the WST-8 assay. Asterisks indicate significantly different from control.
In the canonical inflammasome pathway, ASC and NLRP3 are required to activate caspase-1. ASC was expressed in BMM and J774A.1, while ASC was not expressed in RAW264.7 and was only faintly expressed in J774.1 cells. NLRP3 was expressed almost equally in all four cell types (Fig. 2a). Both the active form of caspase-1 and the active form of IL-1β were released into the culture medium when LPS-primed BMM was stimulated with ATP as was observed in J774A.1 cells (Fig. 2b). It should be noted that most IL-1β released in the culture medium was an active form (17 kDa) in BMM (Fig. 2b), while a larger amount of pro-IL-1β was released than the active form of IL-1β in the culture medium in J774A.1 cells (Fig. 1a). The canonical inflammasome pathway appeared to be activated when ATP was replaced with FTiO2 (Fig. 2b).

a Expression of ASC and NLRP3 in J774.1, RAW264.7, BMM, and J774A.1 cells. The protein levels were measured by immunoblot analysis. The amount of protein for BMM loaded to the gel was reduced to one fifth of the other samples since the ASC signal of BMM was much higher than that of the other cells. b Immunoblot analysis for the detection of IL-1β, IL-6, and caspase-1 in the cell lysate and the culture medium of BMM. The cells were exposed to 5 mM ATP for 3 h alone or at the end of 6-h exposure to 100 ng/mL of LPS. In a separate experiment, the cells were exposed to 100 μg/mL of FTiO2 alone or 100 μg/mL of FTiO2 with 100 ng/mL of LPS for 16 h. Prior to analysis, the culture medium supernatant was concentrated 50 times.
Next, we investigated synergistic effects of LPS and FTiO2 on J774.1 and RAW264.7 cells to see how these cells release inflammatory cytokines without the NLRP3 inflammasome activation. When the cells were co-stimulated with LPS and FTiO2, a remarkable IL-1β release was observed only at cytotoxic concentration of FTiO2 (100 μg/mL, Fig. 3a, b). The release of IL-1β in the culture medium was more efficient when the cells were pre-treated with FTiO2 and then stimulated with LPS than when the cells were stimulated with LPS first and then treated with FTiO2 (Fig. 3c, d).

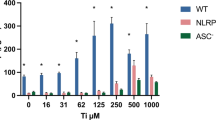
Cytotoxicity of FTiO2 (a), effects of different doses of FTiO2 on LPS-induced IL-1β production (b), effects of pre-treatment of the cells with FTiO2 on LPS-induced IL-1β production (c), and effects of post-treatment with FTiO2 on LPS-primed IL-1β production (d) in the culture medium of J774.1 cells. (a) The cells were treated with various concentrations of FTiO2 for 24 h. Viable cells were then evaluated using the WST-8 assays. Data are presented as means ± SEM (N = 4). Asterisks indicate significantly different from the control group. (b) The cells were treated with (closed columns) or without (hatched columns) 100 ng/mL of LPS in the presence of 0, 10, or 100 μg/mL of FTiO2 for 24 h. IL-1β in the culture medium was then assayed using ELISA. Data are presented as means ± SEM (N = 4). Two-way ANOVA with Tukey’s HSD test indicated the following significant differences: main (p < 0.05); LPS, FTiO2 (0 or 10 versus 100 μg/mL), interaction (p < 0.05), LPS × FTiO2. (c) The cells were treated with (gray columns) or without (open columns) 100 μg/mL of FTiO2 for 24 h. The cells were then washed twice with culture medium and further cultured with 0, 10, or 100 ng/mL of LPS for 24 h following which IL-1β in the culture medium was assayed as in (b). Data are presented as means ± SEM (N = 4). Both the main effects and the interaction were significantly different by two-way ANOVA with Tukey’s HSD test. (d) The cells were treated with 0, 10, or 100 ng/mL of LPS for 24 h. The cells were washed twice with culture medium and were then further cultured in the presence (closed columns) or absence (open columns) of 100 μg/mL FTiO2 for 24 h following which IL-1β in the culture medium was assayed as in (b). Data are presented as means ± SEM (N = 4). The main effects were significantly different by two-way ANOVA (Tukey’s HSD test).
IL-1β and IL-6 mRNA levels were measured in J774.1 cells after exposure to saline (control), LPS, or FTiO2 alone, and the co-exposure to LPS and FTiO2. Both IL-1β and IL-6 mRNA levels of LPS-exposed cells were further increased by co-exposure to FTiO2 (Fig. 4). Figure 5 shows results with immunoblot analyses of IL-1β, IL-6, and caspase-1 in both the cell lysate and culture medium of J774.1 cells following exposure to LPS or LPS plus FTiO2. IL-6 was not retained in the cells and was released in the culture medium. Both protein and mRNA levels of IL-6 were increased by LPS and further increased by co-stimulation of LPS and FTiO2. Pro-IL-1β was not cleaved by co-stimulation of LPS and FTiO2 and most of pro-IL-1β remained inside the cells. The released active form of IL-1β was neither absorbed by the cells nor adsorbed by FTiO2 (Supplementary Fig. 2). FTiO2 appeared to decrease the LPS-induced intracellular pro-IL-1β slightly and increase the pro-IL-1β release in the culture medium, while the mRNA level was further increased by co-exposure with FTiO2. Pro-caspase-1 was not activated by co-stimulation with LPS plus FTiO2 and remained inside the cells. A cytokine array analysis also indicated that the IL-1β release in the culture medium was synergistically increased by co-exposure to LPS and FTiO2 while the synergistic effect of IL-6 release was not observed in J774.1 cells (Supplementary Fig. 3). TCP was not cytotoxic over the concentration range from 100 to 500 μg/mL, and the LPS-induced IL-1β release was only slightly increased by TCP (Supplementary Fig. 4). Caspase-1 activity within the cells was slightly decreased rather than increased following exposure to either FTiO2, LPS, or both in J774.1 (Fig. 6a), suggesting that caspase-1 was not involved in IL-1β release following exposure to either FTiO2 or LPS. Treatment of the cells with either a pan-caspase (z-VAD-FMK) or a caspase-1 inhibitor (z-YVAD-FMK) reduced the IL-1β concentration in the culture medium of J774.1 cells (Fig. 6b), suggesting that caspase-1 may be involved in the IL-1β release although the active form of caspase-1 was not detected by immunoblot analysis.

Changes in mRNA levels of IL-1β (a) and IL-6 (b) following exposure of J774.1 cells to either 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 12 h. Each mRNA level was normalized by that of GAPDH. Data are presented as means ± SEM (N = 3). Asterisks indicate significantly different from the other groups. Number sign indicates significantly different from the control and FTiO2 groups.

Immunoblot analysis for the detection of IL-1β, IL-6, and caspase-1 in J774.1 cells. The cells were exposed to either 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 16 h. Prior to analysis, the culture medium (5 mL) was centrifuged to remove cell debris, and the supernatant was concentrated 50 times. The mature IL-1β (17 kDa) and the cleaved caspase-1 (10 kDa) were not visible. Quantification of the blots is shown on the right in which data are presented as means ± SEM (N = 3). Asterisks indicate significantly different from the control. Number signs indicate significantly different from both the control and LPS groups (Tukey’s post-hoc test).

Effect of LPS and/or FTiO2 stimulation on caspase-1 activity within J774.1 cells (a), and the effect of caspase inhibitors on IL-1β release from LPS plus FTiO2-stimulated J774.1 cells (b). (a) J774.1 cells were treated with either 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 24 h, following which the cells were lysed and centrifuged, and caspase-1 activity in the supernatant was measured. (b) J774.1 cells were treated with 100 μg/mL FTiO2 plus 100 ng/mL LPS in the presence or absence of 2 μM z-VAD-FMK (V) or 10 μM z-YVAD-FMK (Y) for 24 h following which IL-1β in the culture medium was assayed using ELISA. Data are presented as means ± SEM (N = 4). Asterisks indicate significantly different from control. Number sign indicates significantly different from the other groups.
Finally, we measured of IL-1β, IL-6, and caspase-1 in both the cell lysate and culture medium of RAW264.7 cells following exposure to LPS or LPS plus FTiO2 by immunoblot analysis. The induction and the cell lysate/culture medium distribution patterns of IL-1β, IL-6, and caspase-1 following exposure to LPS or LPS plus FTiO2 in RAW264.7 cells were close to those in J774.1 cells (Fig. 7). These results suggest that the active form of IL-1β is not formed without a sufficient amount of ASC because of the lack of caspase-1 activation, and the concentration of IL-1β in the culture medium measured by ELISA or the cytokine array may reflect that of pro-IL-1β rather than the active form of IL-1β when J774.1 and RAW264.7 are used. It should be noted that induction of pro-IL-1β by LPS was faster than that of IL-6 in RAW264.7 cells.

Immunoblot analysis of caspase-1, IL-1β, and IL-6 in RAW264.7 cells. The cells were exposed to 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 3 or 16 h in 5 mL of OPTI-MEM I medium. Prior to analysis, the culture supernatant was concentrated 50 times using an ultrafiltration membrane (MW, 3000). The cells were lysed with 100-μL RIPA buffer.
DISCUSSION
In the canonical NLRP3 inflammasome pathway, pro-IL-1β is first produced via NF-κB activation that is induced by LPS or pathogen-associated molecular patterns (PAMPs). Subsequently, asbestos and crystalline silica destabilize lysosomes and lead to the activation of caspase-1, and the following processing of pro-IL-1β into its active form. Finally, mature IL-1β is released from macrophages. It is also known that extracellular ATP transmits a danger signal through the purinergic P2X7 receptor and activates caspase-1 [13, 15]. Activated macrophages release ASC specks (1–3 μm) that act as an endogenous danger signal and amplify inflammatory signals to the surrounding cells. Extracellular ASC specks are able to recruit and activate both pro-caspase-1 and pro-IL-1β [4, 16]. However, neither pro-caspase-1 nor pro-IL-1β was cleaved into its active form in J774.1 and RAW264.7 cells in response to co-exposure to LPS and ATP or FTiO2. In contrast to these murine macrophage cell lines, mature IL-1β was released from J774A.1 cells and BMM via the canonical inflammasome pathway in the current study. These results indicate that both J774.1 and RAW264.7 lack the canonical inflammasome pathway, which is consistent with the results of a previous study that active forms of caspase-1 and mature IL-1β (17 kDa) were not formed following stimulation of LPS-primed RAW264.7 cells with ATP [14]. Indeed, ASC was not expressed in RAW264.7 cells and was only faintly expressed in J774.1 cells as determined by immunoblot analysis in the present study (Fig. 2a). It has been reported that gram-positive bacteria but not gram-negative bacteria rapidly activate caspase-1 in mouse primary macrophages and RAW264.7 cells [5], suggesting that activation of caspase-1 is not exclusively carried out via the canonical inflammasome pathway in macrophages.
We previously reported that the cytotoxicity of FTiO2 was much higher than that of spherical TiO2, and that Krox-20 mRNA, which is increased rapidly in response to cellular adhesion to the plastic dish surface, was upregulated in response to FTiO2 in rat macrophages [17, 18]. High aspect ratio zeolites were also reported to be cytotoxic [19]. Fibrous and refractory particles are recognized as danger signals by macrophages probably through incomplete phagocytosis that is also called “frustrated phagocytosis” [20] or through subsequent damage to lysosomes [3]. The present study showed that, among 40 common cytokines tested, only the IL-1β level was synergistically increased in the culture medium of J774.1 cells following co-exposure to LPS and a cytotoxic concentration of FTiO2 (Supplementary Fig. 3). Most extracellular pro-IL-1β was released from damaged cells as the IL-1β release was observed only at cytotoxic concentrations of ATP and FTiO2. However, the co-exposure to TCP slightly increased the LPS-inducible IL-1β (pro-IL-1β) release at non-toxic concentrations (Supplementary Fig. 4), suggesting that cell damage is not requisite for a slight increase in the IL-1β release.
It has been reported that the IL-1β level in bronchoalveolar lavage fluid was not increased by inhalational exposure to SiO2 or TiO2. However, alveolar macrophages obtained from rats that were intratracheally instilled with those particles produced a significantly higher amount of IL-1β when stimulated with LPS and, in this ex vivo experiment, SiO2 was more potent than TiO2 [21, 22]. Since an antibody raised against mature IL-1β also naturally reacts with pro-IL-1β, the IL-1β measured using ELISA and the membrane array reflect both mature and pro-IL-1β in either in vitro or in vivo experiments. The present study suggest that it is important to measure pro-IL-1β and the active form of IL-1β separately in order to evaluate the inflammatory responses elicited by particulate substances and chemicals that act as danger signals in phagocytic cells. Contrary to IL-1β, extracellular IL-6 measured by ELISA and membrane array assays appeared to directly reflect inflammatory responses of murine macrophage cell lines and BMM (Figs. 1, 2, 5, and 7).
In conclusion, an active form of IL-1β was released into the culture medium of J774A.1 cells and BMM via the canonical inflammasome pathway, while only pro-IL-1β was released from J774.1 and RAW264.7 cells in response to co-exposure to LPS and FTiO2 or ATP. IL-6 was released into the culture medium readily after production in response to LPS alone in all these cells. The present study warns that measurement of IL-1β by ELISA, and membrane array analyses are not appropriate to evaluate the active form of IL-1β.
REFERENCES
Mantecca, Paride, Francesc Farina, Elisa Moschini, Daniele Gallinotti, Maurizio Gualtieri, Annette Rohr, Giulio Sancini, Paola Palestini, and Marina Camatini. 2010. Comparative acute lung inflammation induced by atmospheric PM and size-fractionated tire particles. Toxicology Letters 198 (2): 244–254.
Sandberg, Wiggo J., Marit Lag, Jørn A. Holme, Bernd Friede, Maurizio Gualtieri, Marcin Kruszewski, Per E. Schwarze, Tonje Skuland, and Magne Refsnes. 2012. Comparison of non-crystalline silica nanoparticles in IL-1beta release from macrophages. Particle and Fibre Toxicology 9: 32.
Dostert, Catherine, Virginie Petrilli, Robin Van Bruggen, Chad Steele, Brooke T. Mossman, and Jürg Tschopp. 2008. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320 (5876): 674–677.
Franklin, Bernardo S., Lukas Bossaller, Dominic De Nardo, Jacqueline M. Ratter, Andrea Stutz, Gudrun Engels, Christoph Brenker, Mark Nordhoff, Sandra R. Mirandola, Ashraf Al-Amoudi, Matthew S. Mangan, Sebastian Zimmer, Brian G. Monks, Martin Fricke, Reinhold E. Schmidt, Terje Espevik, Bernadette Jones, Andrew G. Jarnicki, Philip M. Hansbro, Patricia Busto, Ann Marshak-Rothstein, Simone Hornemann, Adriano Aguzzi, Wolfgang Kastenmuller, and Eicke Latz. 2014. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nature Immunology 15 (8): 727–737.
Sokolovska, Anna, Christine E. Becker, W.K. Ip, Vijay A. Rathinam, Matthew Brudner, Nicholas Paquette, Antonie Tanne, Sivapriya K. Vanaja, Kathryn J. Moore, Katherine A. Fitzgerald, Adam Lacy-Hulbert, and Lynda M. Stuart. 2013. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nature Immunology 14 (6): 543–553.
Broz, Peter. 2015. Immunology: Caspase target drives pyroptosis. Nature 526 (7575): 642–643.
Yazdi, Amir S., Greta Guarda, Nicolas Riteau, Stefan K. Drexler, Aubry Tardivel, Isabelle Couillin, and Jürg Tschopp. 2010. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proceedings of the National Academy of Sciences of the United States of America 107 (45): 19449–19454.
Adachi, Takumi, Kazuhiko Takahara, Jun Taneo, Yasuo Uchiyama, and Kayo Inaba. 2013. Particle size of latex beads dictates IL-1beta production mechanism. PloS One 8 (7): e68499.
Simard, Jean-Christophe, Francis Vallieres, Rafael de Liz, Valerie Lavastre, and Denis Girard. 2015. Silver nanoparticles induce degradation of the endoplasmic reticulum stress sensor activating transcription factor-6 leading to activation of the NLRP-3 inflammasome. The Journal of Biological Chemistry 290 (9): 5926–5939.
Kanno, Sanae, Seishiro Hirano, Shoetsu Chiba, Hiroshi Takeshita, Tamanori Nagai, Meri Takada, Kana Sakamoto, and Toshiji Mukai. 2015. The role of Rho-kinases in IL-1beta release through phagocytosis of fibrous particles in human monocytes. Archives of Toxicology 89 (1): 73–85.
Hamilton, Raymond F., Niangqiang Wu, Dale Porter, Mary Buford, Michael Wolfarth, and Andrij Holian. 2009. Particle length-dependent titanium dioxide nanomaterials toxicity and bioactivity. Particle and Fibre Toxicology 6: 35.
Herzyk, D.A.N.U.T.A.J., James N. Allen, Clay B. Marsh, and Mark D. Wewers. 1992. Macrophage and monocyte IL-1 beta regulation differs at multiple sites: Messenger RNA expression, translation, and post-translational processing. Journal of Immunology 149 (9): 3052–3058.
Englezou, Pavlos C., Simon W. Rothwell, Joseph S. Ainscough, David Brough, Robert Landsiedel, Alexei Verkhratsky, Ian Kimber, and Rebecca J. Dearman. 2015. P2X7R activation drives distinct IL-1 responses in dendritic cells compared to macrophages. Cytokine 74 (2): 293–304.
Pelegrin, Pablo, Consuelo Barroso-Gutierrez, and Annmarie Surprenant. 2008. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. Journal of Immunology 180 (11): 7147–7157.
Kojima, Shuji, Yusuke Negishi, Mitsutoshi Tsukimoto, Takato Takenouchi, Hiroshi Kitani, and Ken Takeda. 2014. Purinergic signaling via P2X7 receptor mediates IL-1beta production in Kupffer cells exposed to silica nanoparticle. Toxicology 321: 13–20.
Baroja-Mazo, Alberto, Fatima Martin-Sanchez, Ana I. Gomez, Carlos M. Martinez, Joaquin Amores-Iniesta, Vincent Compan, Maria Barbera-Cremades, Jordi Yague, Estibaliz Ruiz-Ortiz, Jordi Anton, Segundo Bujan, Isabelle Couillin, David Brough, Juan I. Arostegui, and Pablo Pelegrin. 2014. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nature Immunology 15 (8): 738–748.
Hirano, Seishiro, C.D. Anuradha, and Sanae Kanno. 2000. Transcription of krox-20/egr-2 is upregulated after exposure to fibrous particles and adhesion in rat alveolar macrophages. American Journal of Respiratory Cell and Molecular Biology 23 (3): 313–319.
Hirano, Seishiro, C.D. Anuradha, and Sanae Kanno. 2002. Krox-20/egr-2 is up-regulated following non-specific and homophilic adhesion in rat macrophages. Immunology 107 (1): 86–92.
Fenoglio, Ivana, Antonietta Croce, Francesco Di Renzo, Roberta Tiozzo, and Bice Fubini. 2000. Pure-silica zeolites (porosils) as model solids for the evaluation of the physicochemical features determining silica toxicity to macrophages. Chemical Research in Toxicology 13 (6): 489–500.
Schinwald, Anja, Tanya Chernova, and Ken Donaldson. 2012. Use of silver nanowires to determine thresholds for fibre length-dependent pulmonary inflammation and inhibition of macrophage migration in vitro. Particle and Fibre Toxicology 9: 47.
Driscoll, Kevin E., Robert C. Lindenschmidt, James K. Maurer, Janet M. Higgins, and Gregg Ridder. 1990. Pulmonary response to silica or titanium dioxide: Inflammatory cells, alveolar macrophage-derived cytokines, and histopathology. American Journal of Respiratory Cell and Molecular Biology 2 (4): 381–390.
Driscoll, Kevin E., Robert C. Lindenschmidt, James K. Maurer, Larry Perkins, Mary Perkins, and Janet Higgins. 1991. Pulmonary response to inhaled silica or titanium dioxide. Toxicology and Applied Pharmacology 111 (2): 201–210.
ACKNOWLEDGMENTS
We would like to thank Drs. Przemysław Oberbek, Tomasz Bolek, and Michal Wozniak (Warsaw University of Technology) for providing technical information regarding TCP. This study was partially supported by a Grant-in-Aid for Scientific Research (KAKENHI, #26670341) from Japan Society of the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
We declare that there is no financial conflict of interest in the present study.
Electronic Supplementary Material
Supplementary Fig. 1
The immunoblot of RAW264.7 samples with anti-IL-1β antibody was intensified to visualize faint bands. The putative cleaved IL-1β (21 kDa) was visible in the cell lysate but not in the culture medium in the LPS-exposed RAW264.7 cells only when the signal intensity of the blot was increased. (PPTX 96 kb).
Supplementary Fig. 2
Recovery of IL-1β added to the culture medium. J774.1 cells were treated with or without 100 μg/mL of FTiO2 for 12 h. The culture medium was supplemented or not with IL-1β to a final concentration of 100 pg/ml. After a further 6 h of culture, the supernatant was collected and the concentration of IL-1β was measured. Data are presented as means ± SEM (N = 4). (PPTX 90 kb).
Supplementary Fig. 3
Changes in cytokine levels in the culture medium of J774.1 cells after exposure to either 100 μg/mL of FTiO2, 100 ng/mL of LPS, or both for 24 h. The cytokine concentrations were assayed using a cytokine array. Data are presented as means ± SEM of fold increase versus the control value. Asterisk indicates significantly different from the LPS-treated group (N = 3). (PPTX 5648 kb).
Supplementary Fig. 4
Cytotoxicity of TCP (a) and effects of different doses of TCP on LPS-induced IL-1β production (c and d) in J774.1 cells. (a), The cells were incubated in the presence of the indicated concentration of TCP for 24 h, following which cell viability was assayed using WST-8 assay. (b, c) The cells were treated with (gray columns) or without (open columns) 100 ng/mL LPS for 12 (b) or 24 h (c) in the presence or absence of 100 or 500 μg/mL of TCP, and the concentration of IL-1β in the culture medium was measured using ELISA. Both the main effects and the interaction were significantly different by two-way ANOVA (Tukey’s HSD test) in (b) and (c). Data are presented as means ± SEM (N = 5 for (a), and N = 4 for (b) and (c)). (PPTX 136 kb).
Rights and permissions
About this article

Cite this article
Hirano, S., Zhou, Q., Furuyama, A. et al. Differential Regulation of IL-1β and IL-6 Release in Murine Macrophages. Inflammation 40, 1933–1943 (2017). https://doi.org/10.1007/s10753-017-0634-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0634-1